ABSTRACT
While lifestyle changes such as dietary modification and increased physical activity can be very effective in improving glycemic control, over the long-term most individuals with Type 2 diabetes (T2DM) will require medications to achieve and maintain glycemic control. The purpose of this chapter is to provide the healthcare practitioner with an overview of the existing oral and injectable (non-insulin) pharmacological options available for the treatment of patients with T2DM. Currently, there are ten classes of orally available pharmacological agents to treat T2DM: 1) sulfonylureas, 2) meglitinides, 3) metformin (a biguanide), 4) thiazolidinediones (TZDs), 5) alpha glucosidase inhibitors, 6) dipeptidyl peptidase IV (DPP-4) inhibitors, 7) bile acid sequestrants, 8) dopamine agonists, 9) sodium-glucose transport protein 2 (SGLT2) inhibitors and 10) oral glucagon like peptide 1 (GLP-1) receptor agonists. In addition, glucagon like peptide 1 (GLP-1) receptor agonists, dual GLP-1 receptor and GIP receptor agonists, and amylin can be administered by injection. Medications from these distinct classes of pharmaceutical agents may be used as treatment by themselves (monotherapy) or in a combination of 2 or more drugs from multiple classes with different mechanisms of action. A variety of fixed combinations of 2 agents are available in the US and in many other countries. In this chapter we discuss the administration, mechanism of action, effect on glycemic control, other benefits, side effects, and the contraindications of the use of these glucose lowering drugs.
INTRODUCTION
While lifestyle changes such as dietary modification and increased physical activity can be very effective in improving glycemic control, over the long-term most individuals with Type 2 diabetes (T2DM ) will require medications to achieve and maintain glycemic control (1). The purpose of this chapter is to provide the healthcare practitioner with an overview of the existing oral and injectable (non-insulin) pharmacological options available for the treatment of patients with T2DM. The use of these drugs to treat diabetes during pregnancy, in children and adolescents, and for the prevention of diabetes are discussed in other Endotext chapters (2-4). For information on the management of T2DM and selecting amongst the available pharmacological agents see the chapter by Emily Schroeder in Endotext (5).
Currently, there are ten classes of orally available pharmacological agents to treat T2DM: 1) sulfonylureas, 2) meglitinides, 3) metformin (a biguanide), 4) thiazolidinediones (TZDs), 5) alpha glucosidase inhibitors, 6) dipeptidyl peptidase IV (DPP-4) inhibitors, 7) bile acid sequestrants, 8) dopamine agonists, 9) sodium-glucose transport protein 2 (SGLT2) inhibitors and 10) oral glucagon like peptide 1 (GLP-1) receptor agonists (Table 1) (6-8). In addition, glucagon like peptide 1 (GLP-1) receptor agonists, dual GLP-1 receptor and GIP receptor agonists, and amylin can be administered by injection (Table 2) (6-8).
|
Table 1. Currently Available (USA) Oral Hypoglycemic Drugs to Treat Type 2 Diabetes
|
|
General Class
Compound/Brand Name
|
Generic Available
|
Dose Range
|
Cost
|
|
1st Generation Sulfonylureas
|
|
Chlorpropamide/ Diabinese
|
Yes
|
100-750mg qd
|
Low
|
|
Tolazamide/ Tolinase
|
Yes
|
100mg qd to 500mg bid
|
Low
|
|
Tolbutamide/ Orinase
|
Yes
|
500mg qd to 1000mg tid with meals
|
Low
|
|
Acetohexamide/ Dymelor
|
Yes
|
250mg qd to 750mg bid
|
Low
|
|
2nd Generation Sulfonylureas
|
|
Glyburide (Glibenclamide)/ Diabeta, Glynase
|
Yes
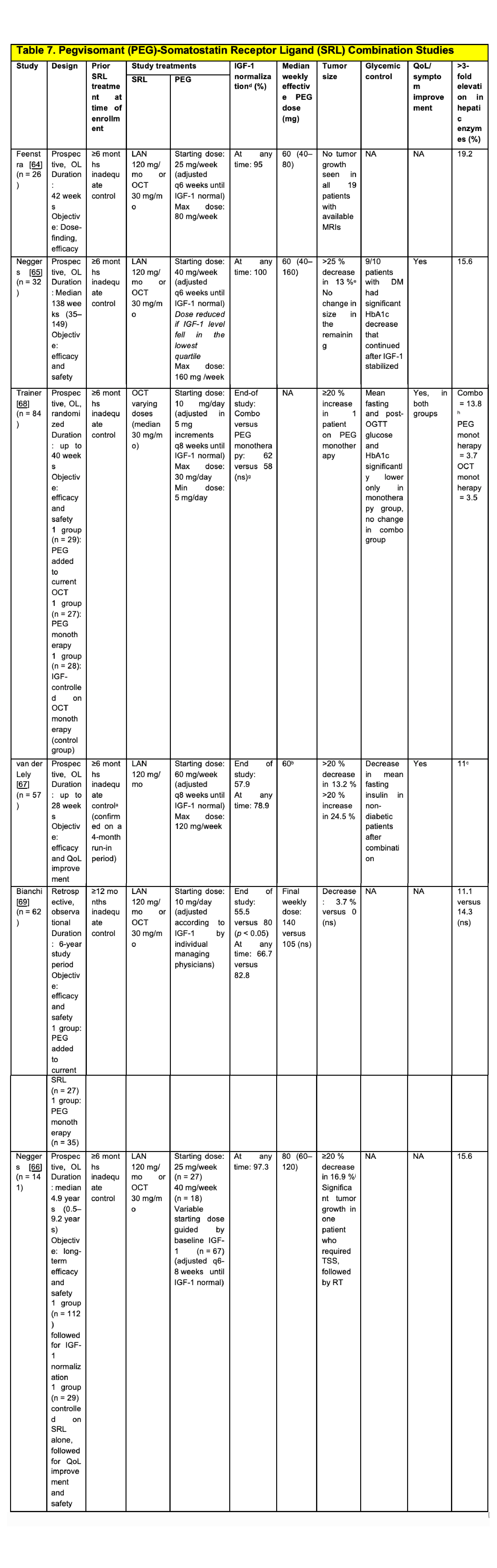
|
2.5mg qd to 10mg bid
|
Low
|
|
Glipizide/ Glucotrol, Glucotrol XL
|
Yes
|
2.5mg qd to 20mg bid
|
Low
|
|
Glimepiride/ Amaryl
|
Yes
|
0.5mg to 8mg qd
|
Low
|
|
Gliclazide/ Diamicron
|
Yes
|
40mg qd to 160mg bid
|
Low
|
|
Meglitinides
|
|
Repaglinide/ Prandin
|
Yes
|
0.5mg to 4 mg with meals. Max 16mg/day
|
Low
|
|
Nateglinide/ Starlix
|
Yes
|
60-120mg tid with meals
|
Low
|
|
Biguanide
|
|
Metformin/ Glucophage, Glucophage XR
|
Yes
|
500-2500mg qd or tid depending upon preparation
|
Low
|
|
Thiazolidinediones (TZDs)
|
|
Rosiglitazone/ Avandia
|
Yes
|
4-8mg qd
|
High
|
|
Pioglitazone/ Actos
|
Yes
|
15-45mg qd
|
Low
|
|
Alpha-glucosidase inhibitors
|
|
Acarbose/ Precose
|
Yes
|
25-100mg tid with meals
|
Low
|
|
Miglitol/ Glyset
|
Yes
|
25-100mg tid with meals
|
High
|
|
Voglibose/ Basen, Voglib
|
Yes
|
0.2mg tid with meals
|
|
|
Dipeptidyl peptidase-IV (DPP-4) inhibitors
|
|
Alogliptin/ Nesina
|
Yes
|
25mg qd
|
High
|
|
Linagliptin/ Tradjenta
|
No
|
5mg qd
|
High
|
|
Sitagliptin/ Januvia
|
No
|
25-100mg qd
|
High
|
|
Saxagliptin/ Onglyza
|
No
|
2.5-5mg qd
|
High
|
|
Vildagliptin/ Galvus
|
No
|
50mg qd
|
|
|
Bile Acid Sequestrant
|
|
Colesevelam/ Welchol
|
No
|
1875mg bid or 3.75-gram packet or bar qd
|
High
|
|
Dopamine Agonist
|
|
Bromocriptine/ Cycloset
|
No
|
0.8 - 4.8mg qAM
|
High
|
|
Sodium-glucose co-transporter-2 (SGLT2) inhibitors
|
|
Canagliflozin/ Invokana
|
No
|
100-300mg qd
|
High
|
|
Dapagliflozin/ Farxiga
|
No
|
5-10mg qd
|
High
|
|
Empagliflozin/ Jardiance
|
No
|
10-25mg qd
|
High
|
|
Ertugliflozin/ Stelgatro
|
No
|
5-15mg qd
|
High
|
|
Glucagon like peptide 1 (GLP-1) receptor agonists
|
|
Semaglutide/ Rybelsus
|
No
|
7-14mg qd
|
High
|
|
Table 2. Currently Available (USA) Injectable Hypoglycemic Drugs to Treat Type 2 Diabetes
|
|
General Class
Compound/Brand Name
|
Generic Available
|
Dose Range
|
Cost
|
|
GLP-1 Receptor Agonist
|
|
Exenatide/ Byetta
|
No
|
5-10mcg bid
|
High
|
|
Exenatide/ Bydureon
|
No
|
2mg once weekly
|
High
|
|
Liraglutide/ Victoza
|
No
|
0.6-1.8mg qd**
|
High
|
|
Albiglutide/ Tanzeum*
|
No
|
30-50mg once weekly
|
High
|
|
Dulaglutide/ Trulicity
|
No
|
0.75-4.5mg once weekly
|
High
|
|
Lixisenatide/ Adlyxin
|
No
|
10-20mcg qd
|
High
|
|
Semaglutide/ Ozempic
|
No
|
0.25-2.0mg once weekly
|
High
|
|
Dual GLP-1 Receptor/GIP Receptor Agonists
|
|
Tirzepatide/ Mounjaro
|
No
|
5mg-15mg once weekly
|
High
|
|
Amylin Mimetic
|
|
Pramlintide/ Symlin
|
No
|
15-120mcg tid with meals
|
High
|
*Withdrawn from market
Medications from these distinct classes of pharmaceutical agents may be used as treatment by themselves (monotherapy) or in a combination of 2 or more drugs from multiple classes with different mechanisms of action (6-8). A variety of fixed combination of 2 agents are available in the US and in many other countries (examples shown in Table 3). There are even combinations that contains 3 drugs (Qternmet XR which contains dapagliflozin, saxagliptin, and metformin and Trijardy XR which contains empagliflozin, linagliptin, and metformin). Additionally, there are combinations of GLP-1 receptor agonists and insulin (Table 3). These combination products may be useful and attractive to the patient, as they provide multiple drugs in a single tablet or injection, offering convenience and increased compliance. In the US, they also enable patients to receive two medications for a single medical insurance co-payment. Most importantly, the addition of a second drug results in an additive improvement in glycemic control. When a patient is on drug A if drug B is added to drug A, there is an improvement in glycemic control. This concept can be extended by the addition of a third drug C, and even a fourth drug D (Figure 1).
|
Table 3. Oral Pharmacological Fixed Combination Therapies to Treat Type 2 Diabetes
|
|
Drug 1
|
Drug 2
|
Brand Name
|
Generic
|
|
Glyburide
|
Metformin
|
Glucovance (discontinued by manufacturer: generic available)
|
Yes
|
|
Glipizide
|
Metformin
|
Metaglip (discontinued by manufacturer; generic available)
|
Yes
|
|
Glimepiride
|
Pioglitazone
|
Duetact
|
Yes
|
|
Glimepiride
|
Rosiglitazone
|
Avandaryl
|
Yes
|
|
Sitagliptin
|
Metformin
|
Janumet
|
No
|
|
Saxagliptin
|
Metformin
|
Kombiglyze XR
|
No
|
|
Pioglitazone
|
Metformin
|
ACTOSplus Met; ACTOSplus Met XR
|
Yes
|
|
Repaglinide
|
Metformin
|
PrandiMet
|
Yes
|
|
Rosiglitazone
|
Metformin
|
Avandamet
|
Yes
|
|
Linagliptin
|
Metformin
|
Jentadueto
|
No
|
|
Alogliptin
|
Metformin
|
Kazano
|
Yes
|
|
Alogliptin
|
Pioglitazone
|
Oseni
|
No
|
|
Canagliflozin
|
Metformin
|
Invokamet
|
No
|
|
Dapagliflozin
|
Metformin
|
Xigduo XR
|
No
|
|
Dapagliflozin
|
Saxagliptin
|
Qtern
|
No
|
|
Empagliflozin
|
Linagliptin
|
Glyxambi
|
No
|
|
Empagliflozin
|
Metformin
|
Synjardy
|
No
|
|
Ertugliflozin
|
Metformin
|
Segluromet
|
No
|
|
Ertugliflozin
|
Sitagliptin
|
Steglujan
|
No
|
|
Lixisenatide
|
Glargine Insulin
|
Soliqua
|
No
|
|
Liraglutide
|
Degludec Insulin
|
Xultophy
|
No
|
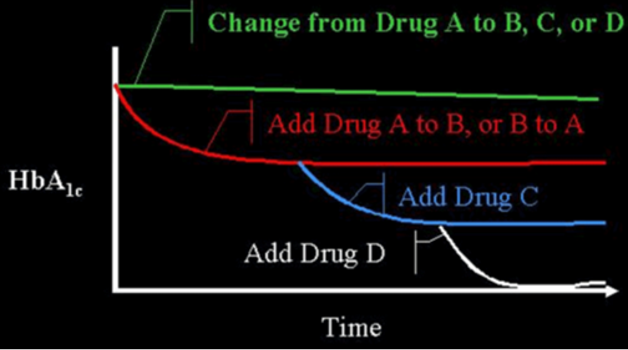
Figure 1. Efficacy When Oral Agents are Used as Add-On Therapy. When a patient is on drug A and they are changed to drug B, C, or D, often no improvement in glucose control will be seen. However, if drug B is added to drug A, there is an improvement. This concept can often be extended by the addition of a third drug (C), or even a fourth drug (D). There is decreasing benefit for each additional drug as the baseline A1c level decreases. Note that there is limited data on the use of 4 drug combinations.
OVERVIEW OF DRUGS
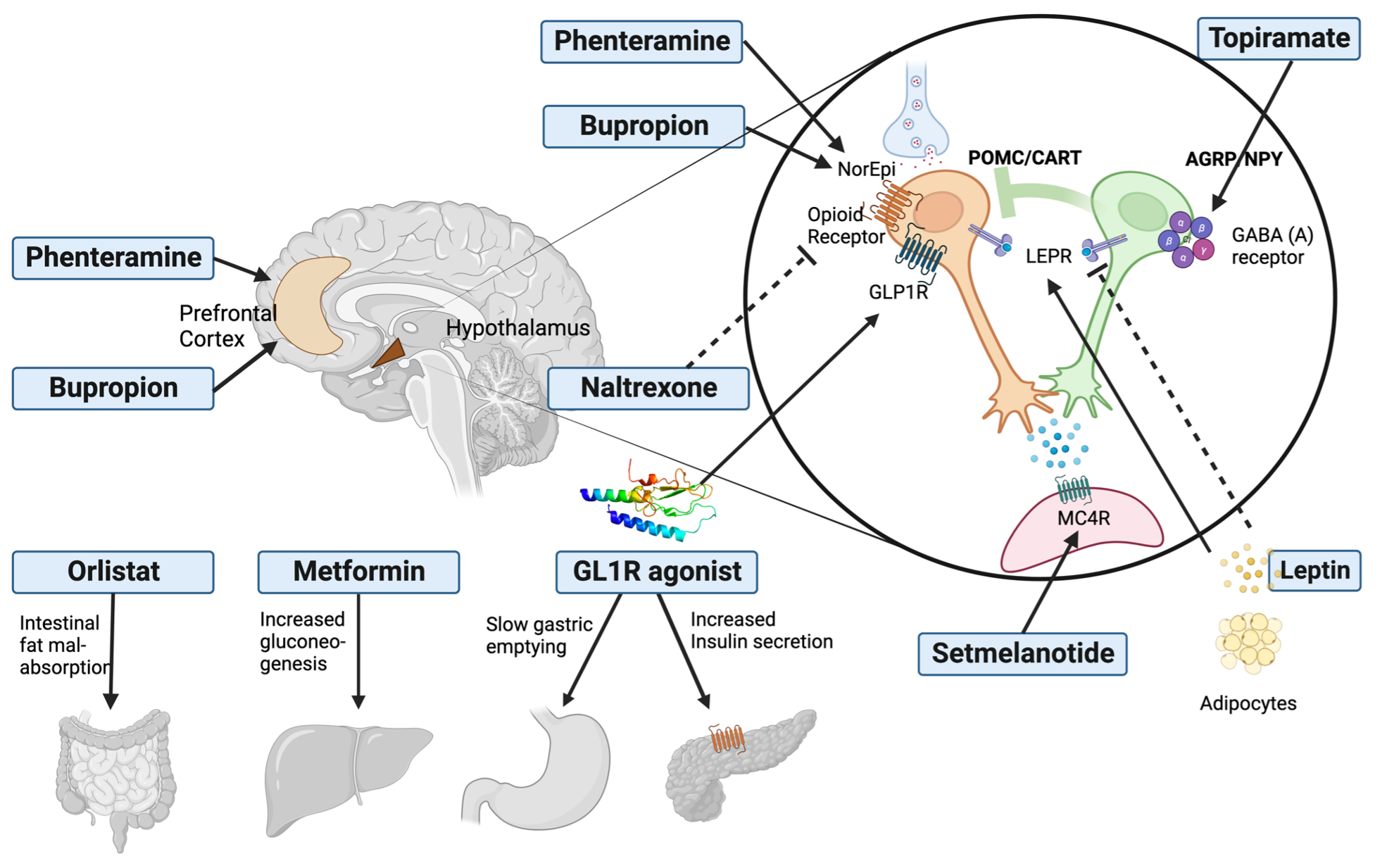
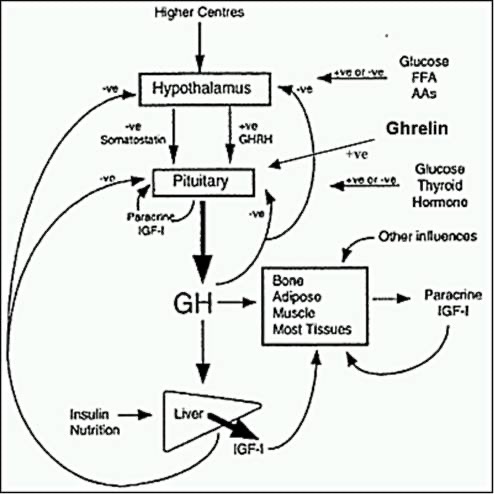
There are a number of different abnormalities that contribute to the hyperglycemia that occurs in patients with T2DM (9). Therefore, the drugs used to treat patients with T2DM can have a number of different mechanisms by which they lower glucose levels. Figure 2 shows the various sites of action of the pharmacological therapies for the treatment of T2DM.
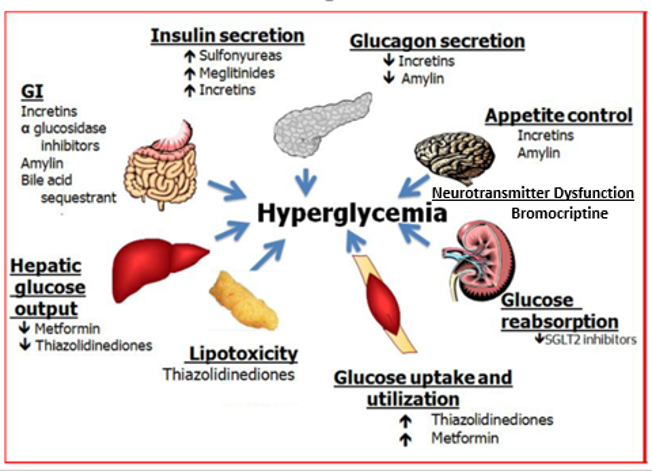
Figure 2. Sites of Action of Pharmacological Therapies for the Treatment of Type 2 Diabetes.
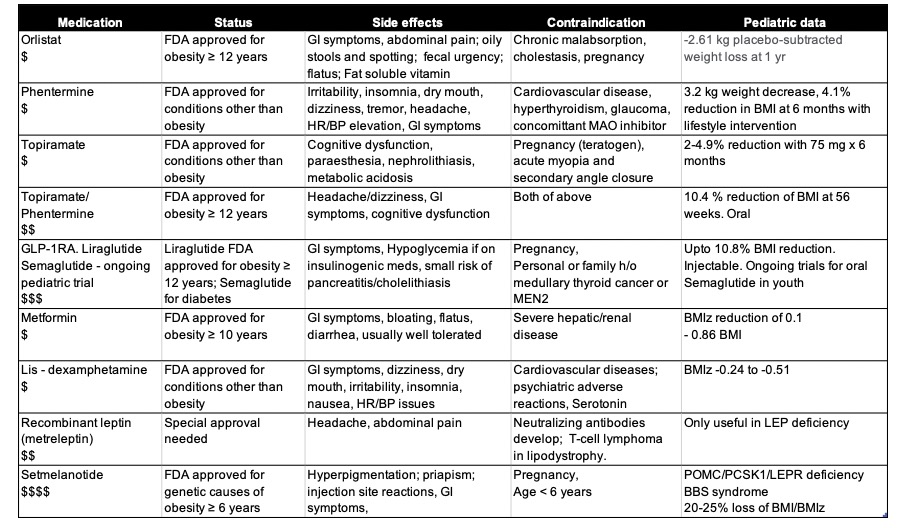
A broad overview of the most commonly used drugs to treat T2DM is shown in Table 4 and the effect of drugs on blood lipid levels is shown in Table 5.
|
Table 4. Benefits and Side Effects of Commonly Used Drugs
|
|
Drugs
|
Ability to Lower Glucose
|
Risk of Hypoglycemia
|
Weight Change
|
Effect on ASCVD
|
Effect on CHF
|
Effect on Renal Disease
|
|
2ndGeneration SU
|
High
|
Yes
|
Increase
|
Neutral
|
Neutral
|
Neutral
|
|
Metformin
|
High
|
No
|
Neutral- modest weight loss
|
Potential Benefit
|
Neutral
|
Neutral
|
|
TZDs
|
High
|
No
|
Increase
|
Potential Benefit (Pioglitazone)
|
Increased
|
Neutral
|
|
DPP-4 inhibitors
|
Intermediate
|
No
|
Neutral
|
Neutral
|
Potential Increase (saxagliptin and alogliptin)
|
Neutral
|
|
SGLT2 inhibitors
|
Immediate
|
No
|
Decrease
|
Potential Benefit
|
Benefit
|
Benefit-
Reduced progression of renal failure
|
|
GLP-1 receptor agonists
|
High
|
No
|
Decrease
|
Benefit
|
Neutral-
Potential Benefit
|
Benefit-
Decreased proteinuria
|
|
Table 5. Effect of Glucose Lowering Drugs on Lipid Levels*
|
|
Metformin
|
Modestly decrease triglycerides and LDL-C
|
|
Sulfonylureas
|
No effect
|
|
DPP4 inhibitors
|
Decrease postprandial triglycerides
|
|
GLP1 analogues
|
Decrease fasting and postprandial triglycerides
|
|
Acarbose
|
Decrease postprandial triglycerides
|
|
Pioglitazone
Rosiglitazone
|
Decrease triglycerides and increase HDL-C. Small increase LDL-C but a decrease in small dense LDL
|
|
SGLT2 inhibitors
|
Small increase in LDL-C and HDL-C
|
|
Colesevelam
|
Decrease LDL-C. May increase triglycerides
|
|
Bromocriptine-QR
|
Decrease triglycerides
|
|
Insulin
|
No effect
|
*These effects are beyond benefits of glucose lowering
Bloomgarden et al reported results from a meta-regression analysis of 61 clinical trials evaluating the efficacy of the five major classes of oral anti-hyperglycemic agents (10). The results demonstrated that there is a strong direct correlation between baseline A1c level and the magnitude of the decrease in fasting glucose and A1c induced by these drugs (i.e., significantly greater reductions in both fasting plasma glucose and A1c were observed in groups with higher baseline A1c levels). Thus, expectations for the overall magnitude of effect from a given agent might be modest when treating patients whose baseline A1c is <7.5-8.0% while in patients with elevated A1c levels the effect of drug therapy may be more robust (figure 3). A separate meta-analysis of 59 clinical studies reached similar conclusions (11). These results indicate that comparing efficacies among different anti-diabetic medications is challenging, when the baseline HbA1c is different in the studies being compared.
Additionally, the population of patients studied can impact the efficacy of a particular class of drug. For example, patients with limited beta cell function will have a decreased response to sulfonylurea drugs as these agents work via stimulating insulin secretion by the beta cells while TZDs are most effective in patients with insulin resistance. Another example would be the decrease in efficacy of SGLT2 inhibitors lowering A1c levels in patients with decreased renal function. It is thus very difficult to compare the glucose lowering effects of different hypoglycemic drugs except in direct head-to-head comparison studies.
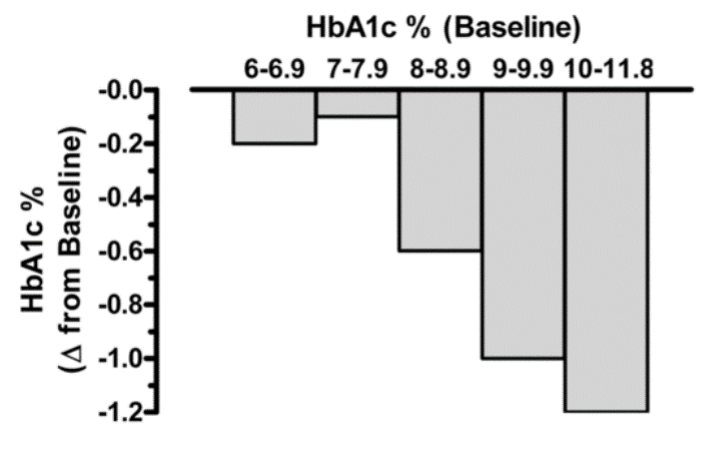
Figure 3. Relationship between baseline A1c level and the observed reduction in A1c with oral anti-hyperglycemic medications. Irrespective of drug class, the baseline glycemic control markedly influences the overall magnitude of efficacy. Data from Bloomgarden et al, Table 1 (10).
A recent model-based meta-analysis was used to compare glycemic control between a large number of drugs adjusted for important differences between studies, including duration of treatment, baseline A1c, and drug dosages (12). In this analysis 229 studies with 121,914 patients were utilized. Table 6 shows the estimated decrease in A1c levels for different drugs in patients that are drug naïve with an A1c of 8% and a weight of 90kg after 26 weeks of treatment. If one averages the effect on A1c of the highest doses for each drug in a specific drug class the reductions in A1c for each class of drug are metformin 1.09%, sulfonylureas 1.0%, TZDs 0.95%, DPP-4 inhibitors 0.66%, SGLT2 inhibitors 0.83%, and GLP-1 receptor agonists 1.24%. These data and the individual data for each drug in table 6 provides a rough estimate of the efficacy of various drugs and drug classes in lowering A1c levels. One should note that within a drug class there may be differences in the ability of different drugs to lower A1c levels. This is particularly true with the GLP-1 receptor agonist drugs. For additional information there is a website that provides updated comparisons of various agents to treat patients with T2DM (https://www.comparediabetesdrugs.com/). Figures 4, 5, and 6 show the effect of glucose lowering drugs on A1c levels, change in weight, and hypoglycemia (graphs from https://www.comparediabetesdrugs.com/ June 28, 2020).
|
Table 6. Estimated Efficacy of Hypoglycemic Drugs Available in US (13)
|
|
Drug
|
A1c % Decrease
|
Drug
|
A1c % Decrease
|
|
Metformin 2000mg
|
1.01
|
Dulaglutide 0.75
|
1.18
|
|
Metformin 2550mg
|
1.09
|
Dulaglutide 1.5mg
|
1.36
|
|
Glipizide 5-20mg
|
0.86
|
Exenatide 10ug BID
|
0.86
|
|
Glyburide 1.25-20mg
|
1.17
|
Exenatide 2mg QW
|
1.16
|
|
Glimepiride 1-8mg
|
0.97
|
Exenatide 2mg QWS
|
1.14
|
|
Pioglitazone 15mg
|
0.62
|
Liraglutide 0.6mg
|
0.88
|
|
Pioglitazone 30mg
|
0.85
|
Liraglutide 1.2mg
|
1.13
|
|
Pioglitazone 45mg
|
0.98
|
Liraglutide 1.8mg
|
1.25
|
|
Rosiglitazone 4mg
|
0.67
|
Lixisenatide 10ug
|
0.44
|
|
Rosiglitazone 8mg
|
0.91
|
Lixisenatide 20ug
|
0.66
|
|
Canagliflozin 100mg
|
0.84
|
Semaglutide 0.5mg
|
1.43
|
|
Canagliflozin 300mg
|
1.01
|
Semaglutide 1.0mg
|
1.77
|
|
Dapagliflozin 5mg
|
0.65
|
Alogliptin 12.5mg
|
0.58
|
|
Dapagliflozin 10mg
|
0.73
|
Alogliptin 25mg
|
0.66
|
|
Empagliflozin 10mg
|
0.69
|
Linagliptin 5mg
|
0.59
|
|
Empagliflozin 25mg
|
0.77
|
Saxagliptin 2.5mg
|
0.59
|
|
Ertugliflozin 5mg
|
0.73
|
Saxagliptin 5mg
|
0.67
|
|
Ertugliflozin 15mg
|
0.81
|
Sitagliptin 100mg
|
0.72
|
The decreases in A1c are modeled for drug naïve patients with an A1c of 8% and a weight of 90kg after 26 weeks of treatment.
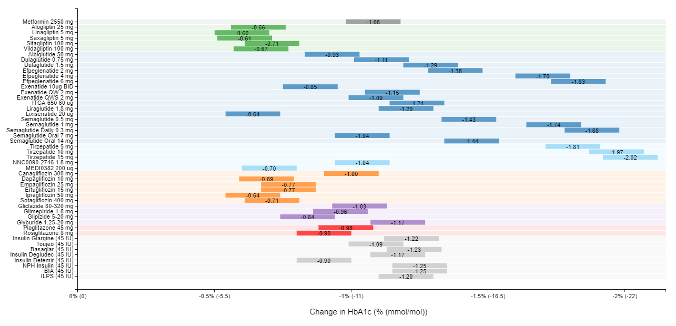
Figure 4. The Effect of Hypoglycemic Drugs on A1c Levels
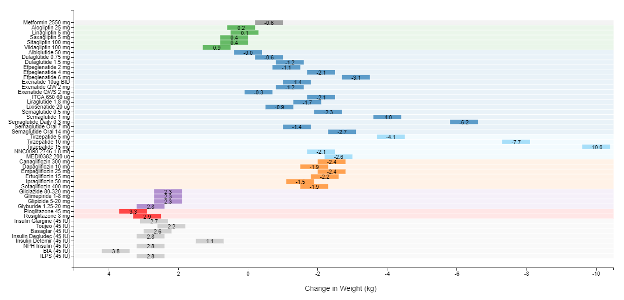
Figure 5. Change in Weight Induced by Hypoglycemic Drugs
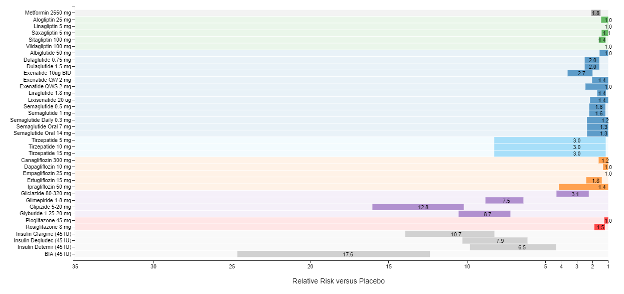
Figure 6. Relative Risk of Hypoglycemia versus Placebo
The NIH is carrying out a study, Glycemia Reduction Approaches in Diabetes: A Comparative Effectiveness (GRADE) Study, that is randomizing approximately 5,000 patients on metformin therapy to sulfonylureas, DPP-4 inhibitors, GLP-1 receptor agonists, and insulin (13). The primary outcome is the time to primary failure defined as an A1c ≥ 7% over an anticipated mean observation period of 4.8 years (range 4-7 years). Preliminary results of this study were presented at the American Diabetes Association meeting (June 2021) and the results as expected demonstrated that the GLP-1 receptor agonist liraglutide was more effective than the sulfonylurea glimepiride and the DPP4 inhibitor sitagliptin in maintaining the A1c < 7% (GLP1 receptor agonist better than sulfonylurea better than DPP-4 inhibitor). It should be noted that the SGLT2 inhibitor and TZD drugs were not included in this study.
SULFONYLUREAS
Introduction
Sulfonylureas were developed in the 1950s and have been widely used in the treatment of patients with T2DM (14,15). First generation sulfonylureas (acetohexamide, chlorpropamide, tolazamide, and tolbutamide) possess a lower binding affinity for the ATP-sensitive potassium channel, their molecular target (vide infra), and thus require higher doses to achieve efficacy (see table 1) (14,15). These first-generation sulfonylureas are currently rarely used. Subsequently, in the 1980s 2nd generation sulfonylureas including glyburide (glibenclamide), glipizide, gliclazide, and glimepiride were developed and are now widely used (14). The 2nd generation sulfonylureas are much more potent compounds (~100-fold). Sulfonylureas can be used as monotherapy or in combination with any other class of oral diabetic medications except meglitinides because they lower glucose levels by a similar mechanism of action (14,16).
Key characteristics of the different sulfonylureas are shown in Table 7 (14). Of clinical importance is the duration of action, which varies with the rate of hepatic metabolism and the hypoglycemic activity of the metabolites. Drugs with a long duration of action are more likely to cause severe and prolonged hypoglycemia whereas short acting drugs need to be given multiple times per day (14). Additionally, drugs that are metabolized to active agents (for example glyburide) are also more likely to cause hypoglycemia (14). Most sulfonylureas are metabolized in the liver and are to some extent excreted by the kidney; therefore, hepatic and/or renal impairment increases the risk of hypoglycemia (14).
|
Table 7. Key Characteristics of Sulfonylureas
|
|
Drug
|
Duration of action
|
Metabolites
|
Excretion
|
|
Tolbutamide
|
6–12 h
|
Inactive
|
Kidney
|
|
Chlorpropamide
|
60 h
|
Active or unchanged
|
Kidney
|
|
Tolazamide
|
12–24 h
|
Inactive
|
Kidney
|
|
Glipizide
|
12–24 h
|
Inactive
|
Kidney 80%
Feces 20%
|
|
Glipizide ER
|
>24 h
|
Inactive
|
Kidney 80%
Feces 20%
|
|
Glyburide
|
16–24 h
|
Inactive or weakly active
|
Kidney 50%
|
|
Micronized glyburide
|
12-24 h
|
Inactive or weakly active
|
Kidney 50%
Feces 50%
|
|
Glimepiride
|
24 h
|
Inactive or weakly active
|
Kidney 60%
Feces 40%
|
Administration
Sulfonylureas should be taken 30 minutes before meals starting with a low dose with an increase in dosage until desired glycemic control has been achieved. In patients with a high risk of severe hypoglycemia a very low-dose can be the initial therapy while in patients with very high A1c levels one can initiate therapy at a higher dose.
The recommended starting dose of glipizide is 5 mg approximately 30 minutes before breakfast. Geriatric patients or those with liver or renal disease or other risk factors for severe hypoglycemia can be started on 2.5 mg. Patients with very high A1c levels may be started on a higher dose. Based on the glucose response the dose can be increased weekly by 2.5-5 mg. If a once-a-day dose is not satisfactory or the patient requires more than 15 mg per day one can give the drug before breakfast and dinner. The maximum daily dose is 40 mg per day.
The usual starting dose of extended-release glipizide is 5 mg per day with breakfast. Those patients who are at high risk of hypoglycemia may be started at a lower dose. The dose can be increased based on glucose or A1c measurements. The maximum dose is 20 mg per day.
The usual starting dose of glyburide is 2.5 to 5 mg daily with breakfast or the first main meal. Patients at high risk for hypoglycemia should be started on 1.25 mg per day. The dose should be increased weekly by 2.5 mg based on the glucose response. The maximum dose per day is 20 mg.
The usual starting dose of micronized glyburide is 1.5 to 3 mg daily with breakfast or the first main meal. Patients at high risk for hypoglycemia should be started on 0.75 mg per day. The dose should be increased weekly by 1.5 mg based on the glucose response. The maximum dose per day is 12 mg.
The recommended starting dose of glimepiride is 1 or 2 mg once daily. Patients at increased risk for hypoglycemia should be started on 1 mg once daily. The dose should be increased every 1-2 weeks in increments of 1 or 2 mg based upon the patient’s glycemic response. The maximum dose is 8 mg per day.
The recommended starting dose of gliclazide is 40 - 80mg once daily. Patients at increased risk for hypoglycemia should be started on 40 mg once daily. The dose should be increased every 1-2 weeks in increments of 40 or 80 mg based upon the patient’s glycemic response. The maximum dose is 160mg twice a day.
Mechanism of Action
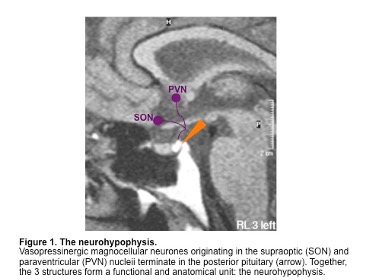
Sulfonylureas are insulin secretagogues and lower blood glucose levels by directly stimulating glucose independent insulin secretion by the pancreatic beta cells (14,16). Through the concerted efforts of GLUT2 (the high Km glucose transporter), glucokinase (the enzyme that phosphorylates glucose), and glucose metabolism, pancreatic beta cells sense blood glucose levels and secrete the appropriate amount of insulin in response (17,18). Glucose metabolism leads to ATP generation and increases the intracellular ratio of ATP/ADP, which results in the closure of the ATP-sensitive potassium channel on the plasma membrane (14,17,19). Closure of this channel depolarizes the membrane and triggers the opening of voltage-sensitive calcium channels, leading to the rapid influx of calcium (14,20). Increased intracellular calcium causes an alteration in the cytoskeleton and stimulates translocation of insulin-containing secretory granules to the plasma membrane and the secretion of insulin (Figure 7) (14).
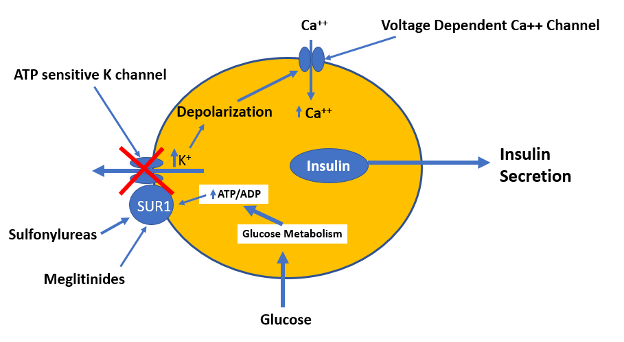
Figure 7. Mechanism by which glucose, sulfonylureas, and meglitinides stimulate insulin secretion by the beta cells.
The KATP channel is comprised of two subunits, both of which are required for the channel to be functional (20). One subunit contains the cytoplasmic binding sites for both sulfonylureas and ATP, and is designated as the sulfonylurea receptor type 1 (SUR1). The other subunit is the potassium channel, which acts as the pore-forming subunit (20). Either an increase in the ATP/ADP ratio or ligand binding by sulfonylureas or meglitinides to SUR1 results in the closure of the KATP channel and insulin secretion (15,20). Studies comparing sulfonylureas and non-sulfonylurea insulin secretagogues have identified several distinct binding sites on the SUR1 that cause channel closure. Some sites exhibit high affinity for sulfonylureas, while other sites exhibit high affinity for meglitinides.
In addition to binding to SUR1, sulfonylureas also bind to Epac2, a protein activated by cAMP (14). Sulfonylurea-stimulated insulin secretion was reduced both in vitro and in vivo in mice lacking Epac2, indicating that Epac2 also plays a role in sulfonylurea induced insulin secretion (21).
In addition to inducing insulin secretion sulfonylureas have other effects that could play a role in lowering blood glucose levels (14). Specifically, sulfonylureas have been shown to decrease hepatic insulin clearance, inhibit glucagon secretion from pancreatic alpha-cells (this may be secondary to increasing insulin secretion), and enhance insulin sensitivity in peripheral tissues (this may be partially due to lowering glucose levels and reducing glucotoxicity) (14). The contribution and importance of these additional effects in mediating the glucose lowering effects of sulfonylureas is uncertain.
Glycemic Efficacy
When used at maximally effective doses, results from well-controlled clinical trials have not indicated a marked superiority of one 2nd generation sulfonylurea over another in improving glycemic control (22). Similarly, 2nd generation sulfonylureas exhibit similar clinical efficacy compared to the 1st generation agents (22). Sulfonylureas do not have a linear dose-response relationship and the majority of the A1C reduction occurs at half maximum dosage. The effect of sulfonylureas as monotherapy or when added to metformin therapy on A1c levels varies but typically results in reductions in A1c of approximately 0.50-1.5% (12,15,16,23,24). If A1c levels are very high decreases in the range of 1.5-2.0% may be seen (15,16,22). Patients with a short duration of diabetes with residual beta cell function (high C-peptide levels) are likely to be most responsive to sulfonylurea therapy (22). Overtime many patients on sulfonylureas require additional therapies (secondary failure). In the ADOPT study, after 5 years 34% of the patients on glyburide monotherapy had fasting glucose levels > 180 mg/dl (i.e., secondary failure) (25). Similarly, in the United Kingdom Prospective Diabetes Study (UKPDS), only 34% of patients attained an A1c <7 % at 6 years treated with sulfonylureas (glyburide or chlorpropamide) and this number declined to 24 % at 9 years (14). This lack of durability of sulfonylurea therapy is likely to due to beta cell exhaustion. In addition, the weight gain induced by sulfonylurea therapy may also adversely affect glycemic control.
Other Effects
CARDIOVASCULAR DISEASE
Based on the University Group Diabetes Project (UGDP) all sulfonylureas carry a “black box” warning regarding cardiovascular disease (26,27). However, the U.K. Prospective Diabetes Study Group (UKPDS) studied a large number of newly diagnosed patients with T2DM at risk for cardiovascular disease. In this study improved glycemic control with sulfonylureas reduced cardiovascular disease by approximately 16%, which just missed being statistically significant (p=0.052) (28). In the UKPDS, A1c was reduced by approximately 0.9% and the 16% reduction in cardiovascular disease was in the range predicted based on epidemiological studies. Thus, the reduction in cardiovascular events was likely due to improvements in glycemic control and not a direct benefit of sulfonylurea treatment. In support of this conjecture is that in the UKPDS, insulin treatment resulted in a similar decrease in A1c levels and reduction in cardiovascular events (28). Additionally, a large randomized cardiovascular outcome study (Carolina Study) reported that linagliptin, a DPP-4 inhibitor, and glimepiride, a sulfonylurea, had similar effects on cardiovascular events (hazard ratio 0.98) (29). Taken together these results suggest that sulfonylureas have a neutral effect on cardiovascular disease.
Side Effects
HYPOGLYCEMIA
The major side effect of sulfonylurea treatment is hypoglycemia, which is more likely to occur and is more severe with long- acting sulfonylureas (14,15). In the UKPDS severe hypoglycemia, defined by need for third-party assistance, occurred each year in 0.4–0.6/100 patients treated with a sulfonylurea while non-severe hypoglycemia was seen in 7.9/100 persons treated with a sulfonylurea (30). Other studies have found even higher rates of severe hypoglycemia with 20–40% of patients receiving sulfonylureas having hypoglycemia and severe hypoglycemia (requiring third-party assistance) occurring in 1–7% of patients (16,30). With continuous glucose monitoring 30% of well controlled patients with T2DM had episodes of hypoglycemia that were often asymptomatic and nocturnal (31). Of great concern these hypoglycemic events were associated with EKG changes, particularly QTc prolongation (31). Other studies have also observed a very high rate of hypoglycemia in patients with T2DM treated with sulfonylureas when monitored using continuous glucose monitoring (32).
Hypoglycemia typically occurs after periods of fasting or exercise. In light of this hypoglycemic risk, initiation of treatment with sulfonylureas should be at the lowest recommended dose and the dose slowly increased in patients with modestly elevated A1c levels. Older patients (> age 65) and patients with hepatic or renal disease are more likely to experience frequent and severe hypoglycemic reactions, particularly if the goals of therapy aim for inappropriately tight glycemic control (14). Many clinicians avoid the use of long acting sulfonylureas (glyburide) in these high-risk patients as glyburide has a higher risk of hypoglycemia compared to other sulfonylureas (33).
WEIGHT GAIN
In the UKPDS, sulfonylurea treatment caused a net weight gain of approximately 3 kg, which occurred during the first 3-4 years of treatment and then stabilized (15,28). Other studies have similarly observed weight gain with sulfonylurea treatment (22).
FIRST GENERATION SIDE EFFECTS
Chlorpropamide can induce hyponatremia and water retention due to inappropriate secretion of antidiuretic hormone (ADH) (14). In addition, tolbutamide and chlorpropamide, in certain susceptible individuals, is associated with alcohol-induced flushing (14). Because of an increased risk of side effects 1st generation sulfonylureas are seldom used.
RARE SIDE EFFECTS
Intrahepatic cholestasis and allergic skin reactions, including photosensitivity and erythroderma may rarely occur (Package insert).
Contraindications and Drug Interactions
Sulfonylureas are best avoided in patients with a sulfa allergy who experienced prior severe allergic reactions (Package insert). Otherwise, cross-reactivity between antibacterial and nonantibacterial sulfonamide agents is rare.
In renal failure, the dose of the sulfonylurea agent will require adjustment based on glucose monitoring to avoid hypoglycemia (14). Because it is metabolized primarily in the liver without the formation of active metabolites, glipizide is the preferred sulfonylurea in patients with renal disease (34).
In the elderly long acting sulfonylureas, such as glyburide, glimepiride and chlorpropamide are not recommended (35).
Sulfonylureas can cause hemolytic anemia in patients with glucose 6-phosphate dehydrogenase (G6PD) deficiency and therefore should be used with caution in such patients (Package insert).
Certain drugs may enhance the glucose-lowering effects of sulfonylureas by inhibition of their hepatic metabolism (antifungals and monoamine oxidase inhibitors), displacing them from binding to plasma proteins (coumarins, NSAIDs, and sulfonamides), or inhibiting their excretion (probenecid) (16).
Summary
While the ability of sulfonylureas to improve glycemic control is robust, the risk of hypoglycemia and weight gain reduce the desirability of this drug class. Additionally, the shorter durability of effectiveness is also a limiting factor. In patients at high risk for the occurrence of severe hypoglycemic reactions or in obese patients, using drugs other than sulfonylureas to treat T2DM is indicated if possible. Similarly, in patients with atherosclerotic cardiovascular disease or at high risk for cardiovascular disease or renal disease other hypoglycemic drugs have advantages. Nevertheless, because sulfonylureas are generic drugs and very inexpensive, they continue to be widely used and play a role in the management of patients with T2DM.
|
Table 8. Summary of the Advantages and Disadvantages of Sulfonylureas
|
|
Advantages
|
Disadvantages
|
|
Inexpensive
|
Hypoglycemia
|
|
Rapid acting
|
Weight gain
|
|
Once a day administration possible
|
Limited durability
|
|
Long history of use
|
Need to titrate dose
|
MEGLINATIDES
Introduction
The meglitinides are non-sulfonylurea insulin secretagogues characterized by a very rapid onset and abbreviated duration of action (16,36). Repaglinide (Prandin), a benzoic acid derivative introduced in 1998, was the first member of the meglitinide class. Nateglinide (Starlix) is a derivative of the amino acid D-phenylalanine and was introduced to the market in 2001. Unlike sulfonylureas, repaglinide and nateglinide stimulation of insulin secretion is dependent on the presence of glucose (36,37). As glucose levels decrease, insulin secretion decreases, which reduces the risk of hypoglycemia compared with sulfonylureas.
Meglitinides are rapidly absorbed with maximum serum concentrations generally attained within 1 hour and then quickly metabolized by the liver cytochrome CYP3A4 and CYP2C8 pathways, producing inactive metabolites, resulting in a plasma half-life of around 1 h (16). This rapid onset and short duration of action results in the ability of this class of drugs to predominantly reduce postprandial glucose levels (36). Because of the rapid onset and short duration of action meglitinides are given 1-30 minutes prior to meals. The drug should not be administered if the patient is going to skip the meal.
The pharmacokinetics of meglitinides differ with nateglinide having a faster onset and shorter duration of action than repaglinide (37). Nateglinide stimulates early insulin release faster and to a greater extent than repaglinide with insulin levels returning to baseline levels more rapidly (36,37).
Administration
The recommended starting dose of nateglinide is 120 mg three times per day before meals (1-30 minutes). In patients who are near their glycemic goal when treatment is initiated the recommended starting dose of nateglinide is 60 mg three times per day before meals. The maximum dose of nateglinide is 120 mg three times per day before meals.
The recommended starting dose of repaglinide for patients whose A1c is less than 8% is 0.5 mg before each meal (1-30 minutes). For patients whose A1c is 8% or greater the starting dose is 1 or 2 mg orally before each meal. The patient’s dose should be doubled up to 4mg with each meal until satisfactory glycemic control is achieved (should wait one week between increasing dose). The maximum daily dose is 16 mg per day.
Mechanism of Action
Meglitinides bind to a different site on SUR1 in β cells that is separate from the sulfonylurea binding site (Figure 7) (16,36). The effect of meglitinide binding is similar to the effect of sulfonylureas binding resulting in the closure of the KATP channel leading to cell depolarization and calcium influx resulting in insulin secretion (16,36,37). However, the relatively rapid onset and short duration of action of meglitinides suits their use as prandial glucose-lowering agents (16,36).
Glycemic Efficacy
Studies have shown that A1c reductions are similar to, or slightly less, than those observed with sulfonylurea or metformin treatment when meglitinides are used as monotherapy (16,36). In studies comparing repaglinide monotherapy with sulfonylurea or metformin therapy the decrease in A1c was similar (36,38). In contrast, a study comparing nateglinide with metformin demonstrated that metformin was more effective in lowering A1c levels (39). In a randomized trial comparing repaglinide and nateglinide in patients with T2DM previously treated with diet and exercise, repaglinide was more effective in lowering A1c levels (1.57% vs. 1.04%) (40). While postprandial glucose levels were similar repaglinide was more effective in reducing fasting glucose levels, probably due to its longer duration of action. These clinical findings can be incorporated into clinical decision making. For example, if the main issue for the patient is postprandial hyperglycemia, and fasting glucoses are near normal, an agent, such as nateglinide, that has a limited effect on the fasting glucose would be ideal. However, if one needs reductions in both fasting and postprandial glucose levels a longer acting agent such as repaglinide is a better choice.
Other Effects
CARDIOVASCULAR DISEASE
The Navigator study was a double-blind, randomized clinical trial in 9,306 individuals with impaired glucose tolerance and either cardiovascular disease or cardiovascular risk factors who received nateglinide (up to 60 mg three times daily) or placebo (41). After 5 years, nateglinide administration did not alter the incidence of cardiovascular outcomes suggesting that meglitinides do not have adverse or beneficial cardiovascular effects.
Side Effects
Similar to sulfonylureas, meglitinides can cause hypoglycemia but the risk of severe hypoglycemia is less (16,36,38). The incidence of hypoglycemia is lower with nateglinide than for repaglinide and nateglinide is less likely to cause severe hypoglycemia (16). In one study, the occurrence of symptomatic hypoglycemia was 2% for nateglinide and 7% for repaglinide (37). Weight gain is also a common side effect of meglitinides (approximately 1-3 kg) with nateglinide leading to less weight gain than repaglinide (16,37).
Contraindications and Drug Interactions
Because meglitinides are metabolized by the liver these drugs should be used cautiously in patients with impaired liver function (Package insert).
Drugs that inhibit CYP3A4 (for example ketoconazole, itraconazole and erythromycin) or CYP2C8 (for example trimethoprim, gemfibrozil and montelukast) can result in the increased activity of meglitinides enhancing the risk of hypoglycemia and should be avoided if possible (38).
Summary
Meglitinides can be useful drugs when there is a need to specifically lower postprandial glucose levels (i.e., patients with fasting glucose in desired range but elevated post meal glucose levels). Additionally, because of their short duration of action meglitinides can be useful in patients who eat erratically as this class of drugs can be given only before meals and the duration of action will match the postprandial increase in glucose. The risk of severe hypoglycemia and weight gain is less than sulfonylureas but still must be considered in patients treated with meglitinides. The development of drugs that do not cause weight gain or severe hypoglycemia and lower postprandial glucose levels have resulted in the limited use of meglitinides.
|
Table 9. Summary of the Advantages and Disadvantages of Meglitinides
|
|
Advantages
|
Disadvantages
|
|
Decrease postprandial glucose
|
Hypoglycemia
|
|
Flexible dosing
|
Weight gain
|
|
Relatively inexpensive
|
Frequent dosing
|
|
Short action allowing for missing meals
|
Need to titrate dose
|
Introduction
Metformin (Glucophage) is a synthetic analog of the natural product guanidine (16). Since its initial clinical use over 50 years ago, metformin has surpassed the sulfonylureas as the most widely prescribed oral agent for T2DM throughout the world because of its proven efficacy on glycemic control as monotherapy and in combination with many other available agents (16). The widespread acceptance of metformin evolved after the realization that lactic acidosis was not a major problem in individuals with normal renal function. Phenformin, a structural analog of metformin, was previously withdrawn from the market in many countries due its propensity to induce lactic acidosis (16).
Administration
The usual starting dose of metformin is 500 mg twice a day with meals. After 1-2 weeks the dose can be increased to 1500 mg per day (750 mg twice a day or 500 mg in AM and 1000 mg in PM). After another 1-2 weeks the dose can be increased to 1000 mg twice a day. The slow increase in dosage is to reduce GI side effects and the dose should not be increased if GI side effects are occurring. The maximum dose is 2550 mg per day which can be given as 850 mg three times per day with meals but most patients are treated with 1000 mg twice a day with breakfast and dinner.
The usual starting dose of metformin extended release is 500 mg with the evening meal (largest meal). The dose can be increased by 500 mg weekly depending upon tolerability. The maximum dose is 2000 mg with the evening meal.
Note the dose of metformin may need to be adjusted based on renal function (discussed below).
Metformin should be temporarily discontinued when patients are unable to eat or drink. Metformin is seldom used in hospitalized patients.
Mechanism of Action
Metformin decreases hepatic glucose production and improves hepatic insulin sensitivity but has only a modest impact on peripheral insulin-mediated glucose uptake (i.e., insulin resistance), which is likely due to a reduction in hyperglycemia, triglycerides, and free fatty acid levels (42,43). Hyperinsulinemia is reduced and the decrease in hepatic glucose production results in a decrease in fasting glucose levels (16). In addition, metformin also increases intestinal glucose utilization and stimulates GLP-1 secretion (42,43). Insulin secretion is not increased (16). The cellular and molecular mechanisms that account for these changes are not definitively understood.
LIVER
There are several lines of evidence indicating that the liver plays an important role in metformin’s ability to improve glycemic control (42). In humans and rodents, metformin is concentrated in the liver and blocking the uptake of metformin into the liver in mice prevents the ability of metformin to lower blood glucose levels (42,43). As noted above tracer studies in humans show that metformin lowers hepatic glucose production and increases hepatic insulin sensitivity (42). There are a number of proposed mechanisms by which metformin alters hepatic metabolism (42).
- Metformin inhibits mitochondrial ATP production by inhibition of Complex I of the respiratory chain and/or inhibiting mitochondrial glycerophosphate dehydrogenase, which is required to carry reducing equivalents from the cytoplasm into the mitochondria for re-oxidation (42,43). The decrease in ATP production could decrease hepatic gluconeogenesis (43). This also leads to an increase in AMP.
- Metformin increases hepatic AMP levels and AMP is a potent allosteric inhibitor of fructose 1,6-bisphosphatase, a key enzyme in gluconeogenesis (43). In addition, high AMP levels inhibit adenylate cyclase reducing cyclic AMP formation in response to glucagon, which also decreases glycogenolysis and gluconeogenesis (i.e., decreases glucagon activity) (43). The increase in AMP also activates AMP-activated protein kinase.
- Metformin activates AMP-activated protein kinase, which activates catabolic pathways leading to decreased gluconeogenesis, decreased fatty acid synthesis, and increased fatty acid oxidation (42,43). The changes in fatty acid metabolism are thought to account for the improvement in hepatic insulin sensitivity and the decrease in serum triglyceride levels (42).
- Metformin inhibits glycerol-3-phosphate dehydrogenase increasing the cytosolic redox state resulting in a decreased conversion of glycerol and lactate to glucose (44).
INTESTINE
Several lines of evidence indicate that the intestine plays an important role in explaining metformin’s ability to lower blood glucose levels. First, a decrease in hepatic glucose production can only partially account for the decrease in blood glucose (42). Second, in humans with loss-of-function variants in SLC22A1, which decrease the uptake of metformin into the liver, the ability of metformin to lower A1c levels is not impaired (42). Finally, a delayed-release metformin that is retained in the gut, with minimal systemic absorption, is as effective at lowering blood glucose as the standard metformin formulation in patients with T2DM (42,45). There are a number of proposed mechanisms for how the intestine accounts for the beneficial effects of metformin.
- Metformin increases anaerobic glucose metabolism in the intestine resulting in increased intestinal glucose utilization and decreased glucose uptake into the circulation (42). This is likely due to the inhibition of mitochondrial ATP production described above. The increased utilization of glucose by anaerobic metabolism could contribute to metformin induced weight loss.
- Metformin increases GLP-1 secretion, which could increase insulin secretion and decrease glucagon secretion (42). The increase in GLP-1 could also contribute to the weight loss or weight neutral effects of metformin.
- Metformin alters the intestinal microbiome, which could alter glucose metabolism (42,46).
It is clear that there are multiple potential mechanisms by which metformin can improve glucose metabolism and further studies are required to elucidate the relative importance and contribution of these proposed mechanisms and others yet to be identified.
Glycemic Efficacy
Metformin is often used as the initial therapy in patients with diabetes in conjunction with lifestyle changes (6,7). The typical reduction in A1c with metformin therapy is in the range of 1 to 2.0% (16,47). The decrease in A1c induced by metformin is independent of age, weight, and diabetes duration as long as some residual β-cell function remains (16). One retrospective study has reported that African-Americans have a greater decrease in A1c with metformin compared to Caucasians (48). The effect of immediate release and extended release metformin on A1c levels is similar (49). In head-to-head trials, metformin has been shown to produce equivalent reductions in A1c as sulfonylureas and thiazolidinediones but is more potent than DPP-4 inhibitors (47).
The durability of glycemic control with metformin is more prolonged than with sulfonylureas but shorter than with TZDs (25). After 5 years of monotherapy, 15% of individuals on rosiglitazone therapy, 21% of individuals on metformin therapy, and 34% of individuals on glyburide (glibenclamide) therapy had fasting glucose levels above the acceptable range (25). The ability to maintain an A1c <7% was 57 months with rosiglitazone, 45 months with metformin, and 33 months with glyburide (glibenclamide) (25).
In addition to the ability to improve glycemic control in monotherapy, metformin in combination with sulfonylureas, meglinitides, TZDs, DPP-4 inhibitors, SGLT-2 inhibitors, insulin, and GLP-1 receptor agonists lowers A1c levels and often allows for patients to achieve their A1c goals (47). As shown in Table 3 there are a large number of combination tablets that include metformin with other glucose lowering drugs.
Hypoglycemia does not occur with metformin monotherapy (47). Hypoglycemia may occur with metformin during concomitant use with other glucose-lowering agents such as sulfonylureas and insulin.
Other Effects
WEIGHT
Metformin is weight neutral or can sometimes result in a modest weight loss (up to 4 kg) (47). When used in combination with sulfonylureas or insulin it blunts the weight gain induced by these agents.
LIPIDS
Metformin decreases serum triglyceride levels and LDL-C levels without altering HDL-C (50,51). In a meta-analysis of 37 trials with 2,891 patients, metformin decreased triglycerides by 11.4mg/dl when compared with control treatment (p=0.003) (50). In an analysis of 24 trials with 1,867 patients, metformin decreased LDL-C by 8.4mg/dl compared to control treatment (p<0.001) (50). In contrast, metformin did not significantly alter HDL-C levels (50). It should be noted that in the Diabetes Prevention Program 3,234 individuals with impaired glucose metabolism were randomized to placebo, intensive lifestyle, or metformin therapy (52). In the metformin therapy group no significant changes were noted in triglyceride, LDL-C, or HDL-C levels compared to the placebo group. Thus, metformin may have small effects on lipid levels.
CARDIOVASCULAR DISEASE
In the UKPDS, metformin, while producing a similar improvement in glycemic control as insulin or sulfonylureas, markedly reduced cardiovascular disease by approximately 40% (53). In the ten-year follow-up the patients randomized to metformin in the UKPDS continued to show a reduction in MI and all-cause mortality (54). Two other small randomized controlled trials have also demonstrated cardiovascular benefits with metformin therapy. A study by Kooy et al compared the effect of adding metformin or placebo in overweight or obese patients already on insulin therapy (55). After a mean follow-up of 4.3 years this study observed a reduction in macrovascular events (HR 0.61 CI- 0.40-0.94, p=0.02), which was partially accounted for by metformin’s beneficial effects on weight. In this study the difference in A1c between the metformin and placebo group was only 0.3%. Hong et al randomized non-obese patients with coronary artery disease to glipizide vs. metformin therapy for three years (56). A1c levels were similar, but there was a marked reduction in cardiovascular events in the metformin treated group (HR 0.54 CI 0.30- 0.90, p=0.026). These results suggest that metformin may reduce cardiovascular disease and that this effect is not due to improving glucose control. Metformin decreases weight or prevents weight gain and lowers lipid levels and these or other non-glucose effects may account for the beneficial effects on cardiovascular disease. Larger cardiovascular outcome studies are required to definitively demonstrate a beneficial effect of metformin on cardiovascular disease.
POLYCYSTIC OVARY SYNDROME (PCOS)
In patients with PCOS metformin lowers serum androgen levels, increases ovulations, and improves menstrual frequency (57). Metformin may also be associated with weight loss in some women with PCOS (57). Metformin combined with clomiphene may be the best combination in obese women with PCOS to improve fertility (57). For a detailed discussion of the treatment of PCOS see the chapter on polycystic ovary syndrome in Endotext (57).
CANCER
Multiple epidemiological studies have demonstrated an association between metformin treatment and a reduced cancer incidence and mortality (58,59). Treatment with metformin has been associated with a decreased risk of breast, colon, liver, pancreas, prostate, endometrium and lung cancer and marked reductions in cancer-specific mortality for colon, lung and early-stage prostate cancer and improvements in survival for breast, colon, endometrial, ovarian, liver, lung, prostate and pancreatic cancer (58,59). A wide variety of different mechanisms have been proposed that could account for metformin’s anti-tumor effects providing biological plausibility (59). However, data from large randomized controlled trials have not yet definitively demonstrated whether metformin can prevent the development of cancer or is useful in the treatment of cancer (58-60). Further studies are required to elucidate the potential role of metformin in oncology.
Side Effects
GASTROINTESTINAL
The most common side effects of metformin are diarrhea, nausea, and/or abdominal discomfort, which can occur in up to 50% of patients (16,47). These side effects are usually mild and disappear with continued drug administration. The GI side effects are dose-related and slow titration to allow for tolerance can reduce the occurrence of these symptoms (47). Administrating metformin three times a day with meals instead of twice a day may also reduce GI side effects. A small number of patients cannot tolerate the drug, even at low doses (47). Extended-release metformin [metformin XR]) causes fewer GI symptoms and can be used in patients who do not tolerate immediate release metformin (47).
Studies have shown that reduced function of plasma membrane monoamine transporter or organic cation transporter 1 leads to an increase in metformin GI side effects (61,62). Use of drugs that inhibit organic cation transporter 1 activity (including tricyclic antidepressants, citalopram, proton-pump inhibitors, verapamil, diltiazem, doxazosin, spironolactone, clopidogrel, rosiglitazone, quinine, tramadol and codeine) increased intolerance to metformin (61).
LACTIC ACIDOSIS
A very rare complication of metformin therapy is lactic acidosis (47). This complication was much more common with phenformin therapy, the initial biguanide, and the risk with metformin is estimated to be 20 times less (47). The estimated incidence of metformin-associated lactic acidosis is 3–10 per 100,000 person-years (47). This is a potentially lethal complication of metformin therapy that typically occurs when renal dysfunction results in very high blood metformin levels, which inhibit mitochondrial function resulting in the overproduction of lactate (47). In addition to renal disorders other risk factors for metformin associated lactic acidosis include sepsis, cardiogenic shock, hepatic impairment, congestive heart failure, and alcoholism (47). In some circumstances the lactic acidosis observed in patients treated with metformin may not be due to metformin but rather to underlying clinical disorders such as severe sepsis.
VITAMIN B12 DEFICIENCY
Studies have demonstrated that vitamin B12 malabsorption is a side effect of metformin therapy (47). A randomized controlled trial showed that metformin 850 mg three times per day for over 4 years resulted in a 19% decrease in B12 levels compared to placebo (63). Moreover, 9.9% of patients treated with metformin developed vitamin B12 deficiency (<150 pmol/l) vs only 2.7% in the placebo group (63). The Diabetes Prevention Program Outcomes Study also demonstrated an increased risk of B12 deficiency with long term metformin use (64). It is now recommended that periodic testing of vitamin B12 levels should be considered in patients on long-term metformin therapy, particularly in the setting of anemia or neuropathy (65).
OVULATION AND PREGNANCY
As discussed above in the polycystic ovary section, treatment of premenopausal women with PCOS with metformin may induce ovulation and thereby result in unplanned pregnancies. In premenopausal anovulatory women started on metformin one needs to discuss the need for contraception.
Contraindications and Drug Interactions
Metformin is contraindicated in patients with advanced kidney or liver disease, acute unstable congestive heart failure, conditions marked by decreased perfusion or hemodynamic instability, major alcohol abuse, or conditions characterized by acidosis (47). Metformin therapy should be suspended during serious illness or surgical procedures. Metformin is seldom used in hospitalized patients.
RENAL DISEASE
A major contraindication to the use of metformin is renal disease (47). Metformin is not metabolized and is excreted intact by the kidneys and therefore kidney function is a major determinant of blood metformin levels. eGFR should be obtained prior to initiating therapy. In patients with renal dysfunction or at risk for developing renal dysfunction eGFR should be obtained more frequently. In patients with a eGFR < 30 mL/min/1.73 m2 metformin therapy is contraindicated (47). In patients with an eGFR between 30-60mL/min/1.73 m2 metformin can be used but one should consider using lower doses (47). In patients with eGFR < 45mL/min/1.73 m2 the author typically uses ½ the maximal dose of metformin. In patients with labile renal disease, especially if frequent deteriorations in kidney function occur, metformin is best avoided.
IODINATED CONTRAST STUDIES
FDA guidelines indicate that metformin use should be withheld before iodinated contrast procedures if a) the eGFR is 30–60 mL/min/1.73 m2, b) in the setting of liver disease, alcoholism, or heart failure, or c) if intra-arterial contrast is used. The eGFR should be checked 48 hours later and metformin restarted if renal function remains stable.
DRUG INTERACTIONS
Carbonic anhydrase inhibitors, such as topiramate or acetazolamide, can decrease serum bicarbonate levels and induce a non-anion gap, hyperchloremic metabolic acidosis. Concomitant use of these drugs with metformin may increase the risk for lactic acidosis (Package Insert).
Certain drugs, such as ranolazine, vandetanib, dolutegravir, and cimetidine, may interfere with common renal tubular transport systems that are involved in the renal elimination of metformin and therefore can increase systemic exposure to metformin and may increase the risk for lactic acidosis (Package Insert).
Summary
Metformin is a commonly used as a the first drug for the treatment of diabetes because of excellent efficacy, an outstanding safety profile, low cost, and a long history of use without significant problems.
|
Table 10. Summary of the Advantages and Disadvantages of Metformin
|
|
Advantages
|
Disadvantages
|
|
Inexpensive
|
GI side effects
|
|
No hypoglycemia
|
B12 deficiency
|
|
Once a day administration possible
|
Lactic acidosis (very rare)
|
|
Long history of use
|
Need to monitor renal function
|
|
No weight gain and maybe weight loss
|
|
|
May decrease cardiovascular disease
|
|
THIAZOLIDINEDIONES (TZDS)
Introduction
Troglitazone (Rezulin), pioglitazone (Actos), and rosiglitazone (Avandia) are members of the thiazolidinedione (TZD) class of insulin sensitizing compounds that activate PPAR gamma (16,66). Troglitazone was withdrawn from the US, European, and Japanese markets in 2000 due to an idiosyncratic hepatic reaction leading to hepatic failure and death in some patients (16,66). This idiosyncratic hepatic reaction has not occurred with pioglitazone or rosiglitazone (66). TZDs decrease insulin resistance and thereby enhance the biological response to endogenously produced insulin, as well as exogenous insulin (66).
Administration
Initiate pioglitazone at 15 mg or 30 mg once a day with or without food. Use 15mg in patients where there is concern of fluid retention. If there is inadequate glycemic control, the dose can be increased in 15 mg increments up to a maximum of 45 mg once daily.
Initiate rosiglitazone at 4 mg once a day with or without food. If there is inadequate glycemic control, the dose can be increased to a maximum of 8 mg once daily.
Because the maximum effect of TZDs on glycemic control may take 10-14 weeks one should wait 12 weeks before deciding whether to increase the dose of TZDs.
Mechanism of Action
The primary effect of pioglitazone and rosiglitazone is the reduction of insulin resistance resulting in an improvement of insulin sensitivity (16,66,67). Pioglitazone and rosiglitazone are selective agonists for the PPAR gamma receptor, a member of the super-family of nuclear hormone receptors that function as ligand-activated transcription factors (66,67). In the absence of ligand, PPARs bind as hetero-dimers with the 9-cis retinoic acid receptor (RXR) and a multi-component co-repressor complex to a specific response element (PPRE) within the promoter region of their target genes (66,67). Once PPAR gamma is activated by ligand, the co-repressor complex dissociates allowing the PPAR-RXR heterodimer to associate with a multi-component co-activator complex resulting in an increased rate of gene transcription (66,67). Additionally, PPAR gamma can repress target gene expression by negative feedback on other signal transduction pathways, such as the nuclear factor kB (NF-kB) signaling pathway, in a DNA binding independent manner (66). The target genes of PPAR gamma include those involved in the regulation of lipid and carbohydrate metabolism and inflammation (66,67).
PPAR gamma is highly expressed in adipose tissue while its expression in skeletal muscle is low (66,67). In the liver PPAR gamma expression is low but increases in obesity and thus in obese individuals it is possible that TZDs directly affect the liver (68). It is likely that the primary effects of TZDs are on adipose tissue, followed by secondary benefits on other target tissues of insulin (66). TZDs promote fatty acid uptake and storage in adipose tissue resulting in a decrease in circulating fatty acids and a decrease in fat accumulation in liver, muscle and pancreas leading to the protection of these tissues from the harmful metabolic effects of higher levels of fatty acids (16,66). This decrease in fat accumulation in liver and muscle leads to an improvement in insulin action and the decrease in the pancreas may improve insulin secretion. Additionally, PPAR gamma agonists increase the expression and circulating levels of adiponectin, an adipocyte-derived protein with insulin sensitizing activity (66). A decrease in the gene expression of other adipokines involved in induction of insulin resistance, such as TNF-alpha, resistin, etc. are likely to also contribute to the improvement in insulin resistance that occurs with TZDs (66). Finally, the activation of PPAR gamma in other tissues may contribute to the beneficial effects of TZDs.
Glycemic Efficacy
Pioglitazone and rosiglitazone decrease A1c levels to a similar degree as metformin and sulfonylurea therapy (typically a 1.0-1.5% decrease in A1c) (16,66). The decreases in fasting plasma glucose were observed as early as the second week of therapy but maximal decreases occurred after 10-14 weeks (16,69). This differs from other hypoglycemic drugs where the maximal effect occurs more rapidly. TZDs lower both fasting and postprandial glucose levels (66). TZDs are more effective in improving glycemic control in patients with marked insulin resistance (70).
TZDs are effective in combination with other hypoglycemic drugs including insulin (16,37,69). TZDs do not cause hypoglycemia when used as monotherapy or in combination with metformin (16,37). In combination with insulin or insulin secretagogues, TZDs can potentiate hypoglycemia. If hypoglycemia occurs one needs to adjust the dose of insulin or insulin secretagogues.
The durability of glycemic control with TZDs is more prolonged than with either sulfonylureas or metformin (18). After 5 years of monotherapy, 15% of individuals on rosiglitazone, 21% of individuals on metformin, and 34% of individuals on glyburide (glibenclamide) had fasting glucose levels above the acceptable range (18). The ability to maintain an A1c <7% was 57 months with rosiglitazone, 45 months with metformin, and 33 months with glyburide (glibenclamide) (18). Similar results were observed when pioglitazone therapy was compared to sulfonylurea therapy (71). After 2-years of therapy 47.8% of pioglitazone-treated patients and only 37.0% of sulfonylurea-treated patients maintained an A1c <8%. Studies have shown that TZDs improve and preserve beta cell function, which may account for their better durability (72-74).
Other Beneficial Effects
PROTEINURIA
A meta-analysis of 15 studies (5 with rosiglitazone and 10 with pioglitazone) involving 2,860 patients demonstrated that TZDs decreased urinary albumin excretion in patients without albuminuria, in patients with microalbuminuria, and in patients with proteinuria (75).
BLOOD PRESSURE
TZDs modestly lower BP. In a review of 37 studies TZDs lowered systolic BP by 4.70 mm Hg and diastolic BP by 3.79 mm Hg (76).
LIPIDS
The effect of TZDs on lipids depends on which agent is used. Rosiglitazone increases serum LDL cholesterol levels, increases HDL cholesterol levels, and only decreases serum triglycerides if the baseline triglyceride levels are high [66]. In contrast, pioglitazone has less impact on LDL cholesterol levels, but increases HDL cholesterol levels, and decreases serum triglyceride levels (77). In the PROactive study, a large randomized cardiovascular outcome study, pioglitazone decreased triglyceride levels by approximately 10%, increased HDL-C levels by approximately 10%, and increased LDL-C by 1-4% (78). It should be noted that reductions in the small dense LDL subfraction and an increase in the large buoyant LDL subfraction are seen with both TZDs (77). Treatment with pioglitazone for 12 weeks resulted in a significant increase in the ability of HDL to facilitate the efflux of cholesterol from cells (79).
In a randomized head-to-head trial, it was shown that pioglitazone decreased serum triglyceride levels and increased serum HDL cholesterol levels to a greater degree than rosiglitazone treatment (80,81). Additionally, pioglitazone increased LDL cholesterol levels less than rosiglitazone. In contrast to the differences in lipid parameters, both rosiglitazone and pioglitazone decreased A1c and C-reactive protein to a similar extent. The mechanism by which pioglitazone induces more favorable changes in lipid levels than rosiglitazone is unclear, but differential actions of ligands for nuclear hormone receptors are well described.
CARDIOVASCULAR DISEASE
Studies with pioglitazone have suggested a beneficial effect on cardiovascular disease. The PROactive study was a randomized controlled trial that examined the effect of pioglitazone vs. placebo over a 3-year period in patients with T2DM and pre-existing macrovascular disease (82). With regard to the primary endpoint (a composite of all-cause mortality, non-fatal myocardial infarction including silent MI, stroke, acute coronary syndrome, endovascular or surgical intervention in the coronary or leg arteries, and amputation above the ankle), there was a 10% reduction in events in the pioglitazone group but this difference was not statistically significant (p=0.095). It should be noted that both leg revascularization and leg amputations are not typical primary end points in cardiovascular disease trials and these could be affected by pioglitazone induced edema. When one focuses on standard cardiovascular disease endpoints, the pioglitazone treated group did demonstrate a 16% reduction in the main secondary endpoint (composite of all-cause mortality, non-fatal myocardial infarction, and stroke) that was statistically significant (p=0.027). In the pioglitazone treated group, blood pressure, A1c, triglyceride, and HDL cholesterol levels were all improved compared to the placebo group making it very likely that the mechanism by which pioglitazone decreased vascular events was multifactorial.
The IRIS trial was a multicenter, double-blind trial that randomly assigned 3,876 patients with insulin resistance but without diabetes and a recent ischemic stroke or TIA to treatment with either pioglitazone or placebo (83). After 4.8 years, the primary outcome of fatal or nonfatal stroke or myocardial infarction occurred in 9.0% of the pioglitazone group and 11.8% of the placebo group (hazard ratio 0.76; P=0.007). All components of the primary outcome were reduced in the pioglitazone treated group. Additionally, in the subgroup of patients with “prediabetes” pioglitazone therapy also reduced cardiovascular events (84). Fasting glucose, fasting triglycerides, and systolic and diastolic blood pressure were lower while HDL cholesterol and LDL cholesterol levels were higher in the pioglitazone group than in the placebo group. Although this study excluded patients with diabetes the results are consistent with and support the results of a protective effect of pioglitazone observed in the PROactive study.
In contrast to the above results, a study compared the effect of pioglitazone vs. sulfonylurea on cardiovascular disease and did not observe a reduction in events with pioglitazone treatment (TOSCA.IT) (85). Patients with T2DM (n= 3,028), inadequately controlled with metformin monotherapy (2-3 g per day), were randomized to pioglitazone or sulfonylurea and followed for a median of 57 months. Only 11% of the participants had a previous cardiovascular event. The primary outcome was a composite of first occurrence of all-cause death, non-fatal myocardial infarction, non-fatal stroke, or urgent coronary revascularization and occurred in 6.8% of the patients treated with pioglitazone and 7.2% of the patients treated with a sulfonylurea (HR 0.96; NS). Limitations of this study are the small number of events likely due to low-risk population studied and the relatively small number of participants. Additionally, 28% of the subjects randomized to pioglitazone prematurely discontinued the medication. Thus, the results of this study should be interpreted with caution. Additionally, it should be noted that when patients in this study were analyzed based on the risk of developing cardiovascular disease those at high risk had a marked reduction in events when treated with pioglitazone compared to the sulfonylurea (86).
Further support for the beneficial effects of pioglitazone on atherosclerosis is provided by studies that have examined the effect of pioglitazone on carotid intima-medial thickness. Both the Chicago and Pioneer studies demonstrated favorable effects on carotid intima-medial thickness in patients treated with pioglitazone compared to patients treated with sulfonylureas (87,88). Additionally, in patients with “prediabetes” pioglitazone also slowed the progression of carotid intima-medial thickness (89). Similarly, Periscope, a study that measured atheroma volume by intravascular ultrasonography, also demonstrated less atherosclerosis in the pioglitazone treated group compared to patients treated with sulfonylureas (90).
There are a large number of potential mechanisms by which pioglitazone might reduce cardiovascular disease (Table 11) (74). In addition to altering risk factors pioglitazone has direct anti-atherogenic effects on the arterial wall that could reduce cardiovascular disease (74).
|
Table 11. Effect of Pioglitazone on Cardiovascular Risk Factors
|
|
Cardiovascular Risk Factor
|
Effect of Pioglitazone
|
|
Visceral Obesity
|
Decreases
|
|
Hypertension
|
Lowers BP
|
|
High Triglycerides
|
Lower TG
|
|
Low HDL cholesterol
|
Increases HDL cholesterol
|
|
Small dense LDL
|
Converts small LDL to large LDL
|
|
Endothelial dysfunction
|
Improves
|
|
Hyperglycemia
|
Lowers A1c
|
|
Inflammation
|
Lowers CRP
|
|
PAI-1
|
Lower PAI-1
|
|
Insulin resistance
|
Reduces
|
|
Hyperinsulinemia
|
Lowers insulin levels
|
While the data from a variety of different types of studies strongly suggests that pioglitazone is anti-atherogenic, the results with rosiglitazone are different. Several meta-analyses of small and short-duration rosiglitazone trials suggested that rosiglitazone was associated with an increased risk of adverse cardiovascular outcomes (91,92). However, the final results of the RECORD study, a randomized trial that was specifically designed to compare the effect of rosiglitazone vs. either metformin or sulfonylurea therapy as a second oral drug in those receiving either metformin or a sulfonylurea on cardiovascular events, have been published and did not reveal a difference in cardiovascular disease death, myocardial infarctions, or stroke (93,94). Similarly, an analysis of patients on rosiglitazone in the BARI 2D trial also did not suggest an increase or decrease in cardiovascular events in the patients treated with rosiglitazone (95).
Thus, while the available data indicate that pioglitazone is anti-atherogenic, the data for rosiglitazone suggests a neutral effect. Whether these differences between pioglitazone and rosiglitazone are accounted for by their differential effects on lipid levels or other factors is unknown.
NONALCOHOLIC FATTY LIVER DISEASE (NAFLD) AND NONALCOHOLIC STEATOHEPATITIS (NASH)
Studies have shown that pioglitazone has beneficial effects on NAFLD and NASH (96). In an early study 55 patients with impaired glucose tolerance or T2DM and liver biopsy-confirmed NASH were randomized to pioglitazone 45 mg/day or placebo (97). After 6 months of therapy liver enzymes improved and hepatic fat decreased, measured by magnetic resonance spectroscopy. Moreover, histologic findings improved including steatosis (P=0.003), ballooning necrosis (P=0.02), and inflammation (P=0.008). However, fibrosis was unchanged. A more recent study randomized 101 patients with prediabetes or T2DM and biopsy-proven NASH to pioglitazone 45 mg/day or placebo for 18 months (98). The primary outcome was a reduction of at least 2 points in the nonalcoholic fatty liver disease activity score in 2 histologic categories without worsening of fibrosis. Pioglitazone treatment resulted in 58% of patients achieving the primary outcome vs. only 17% of the placebo group (p<0.001) and 51% had resolution of NASH compared to 19% of the placebo group (p<0.001). Moreover, pioglitazone treatment improved the fibrosis score.
A meta-analysis of 8 randomized controlled trials (5 using pioglitazone and 3 using rosiglitazone) with 516 patients with biopsy-proven NASH reported that TZD treatment was associated with improved advanced fibrosis (OR, 3.15; P = .01), fibrosis of any stage (OR, 1.66; P = .01), and NASH resolution (OR, 3.22; P < .001) (99). Similar results were observed in patients with and without diabetes. Pioglitazone was more effective in improving NASH than rosiglitazone.
These studies demonstrate that pioglitazone has beneficial effects on NAFLD and NASH. Whether this will result in improved clinical outcomes will require additional studies. TZDs are not FDA approved for the treatment of NAFLD or NASH.
POLYCYSTIC OVARY SYNDROME
TZDs by improving insulin sensitivity decrease circulating androgen levels, improve ovulation rates, and improve glucose tolerance in patients with PCOS (57). Small trials have shown some benefit of TZDs for the treatment of infertility, usually in conjunction with clomiphene (57). Concerns regarding toxicity have limited the use of TZDs for the treatment of PCOS but if a patient has diabetes and TZDs are chosen for treating the diabetes one can anticipate beneficial effects on the PCOS.
Side Effects
WEIGHT GAIN
TZDs lead to an increase in body weight of 2 to 3 kg for every 1 percent decrease in A1c levels (66). In some studies patients gained over 4 kg during a 26-week study (66). Weight gain to a similar degree occurred in monotherapy studies and in studies where TZDs were added to metformin, sulfonylureas, or insulin (66). However, in combination with an SGLT2 inhibitor or a GLP-1 receptor agonist the weight gain was blunted or prevented (100,101). In the ADOPT trial weight gain was greater with TZD therapy than with glyburide therapy (2.5 kg over 5 years) (25). The weight gain induced by TZDs is dose related and can be minimized by using low doses (102).
The TZD induced increase in body weight is due to an expansion of the subcutaneous fat depot whereas the mass of visceral fat remains unchanged or even decreases (66). While weight increases, waist circumference typically remains stable. Stimulation of PPAR gamma in subcutaneous adipocytes stimulates lipid accumulation (67). Fluid retention as discussed below may also contribute to the increase in weight.
FLUID RETENTION
Edema has been reported in 3.0 to 7.5% of patients treated with the TZDs compared with 1.0 to 2.5% in patients on placebo or treated with other oral antidiabetic therapy (103). The increase in fluid retention is dose related. The risk of developing edema is greatest when a TZD is used in combination with insulin (103). The occurrence of edema is reduced when a TZD is used in combination with an SGLT2 inhibitor (100).
TZD induced edema responds poorly to treatment with thiazide and loop diuretics but responds to diuretics that effect the distal tubules such as spironolactone, triamterene, and amiloride (102). Additionally, edema improves when TZD treatment is discontinued (103). The increased fluid retention can lead to an increase in plasma volume resulting in a modest decrease in hemoglobin levels (2-4%) (102).
The increase in fluid retention is likely due to TZDs activating PPAR gamma in the renal tubules leading to the increased expression of the epithelial Na(+) channel resulting in the increased resorption of sodium (104). TZDs have been shown to decrease urine sodium excretion and to increase plasma renin and aldosterone levels (105).
CONGESTIVE HEART FAILURE (CHF)
In a meta-analysis of seven studies with a total of 10,040 participants with 641 CHF events, pioglitazone treatment increased the risk of developing CHF by 33% (RR 1.33, 95% CI 1.14–1.54) (106). Another meta-analysis found that pioglitazone was associated with a 51% increased risk of CHF while rosiglitazone was associated with a 173% increase (107). In the RECORD trial, the rosiglitazone group had an increased rate of severe episodes of CHF resulting in hospital admission or death (OR 2.10, p = 0.001) (93). Similarly, in the PROactive trial, the pioglitazone group also had increased rates of CHF (6% vs. 4%, p = 0.007) (82). Patients treated with TZDs have a higher risk for CHF development if they have a history of cardiovascular disease (102). Interestingly, TZD-associated CHF has not been linked with increased mortality (82,108).
Although TZDs are associated with worsening of CHF or CHF development, they are not associated with adverse effects on cardiac function or structure (102). It is thought that the CHF is mainly due to fluid retention rather than TZDs inducing primarily cardiac dysfunction (102).
OSTEOPOROSIS
Large randomized trials have shown that TZDs increase fracture risk, particularly in women. In the ADOPT study, which compared rosiglitazone, metformin, and glyburide, there was no difference in the incidence of fractures in men (109). However, fractures in women at 5 years was increased in the group treated with rosiglitazone (rosiglitazone 15.1%, metformin 7.3%, and glyburide 7.7%) (109). The increase in fractures with rosiglitazone occurred in pre- and postmenopausal women, and were seen predominantly in the lower and upper limbs (109). In the PROactive study there was a higher rate of bone fractures in females treated with pioglitazone vs. placebo (5.1% vs 2.5%) (110). In the RECORD trial upper and distal lower limb fracture rates were increased mainly in women in the rosiglitazone treatment group (93). Hip and femur fracture were not increased with rosiglitazone treatment (93). In the IRIS trial an increased risk of fracture was seen in both males and females (men 9.4% vs 5.2%; HR, 1.83; women 14.9% vs 11.6%; HR, 1.32) (111). In a meta-analysis of 22 randomized controlled trials with 24,544 participants with 896 fracture cases there was a significantly increased incidence of fracture in women (OR=1.94; P<0.001), but not in men (OR=1.02; P=0.83) treated with TZDs (112). The risk of a fracture was similar with rosiglitazone and pioglitazone treatment and appeared to be similar for participants aged <60 years old and older than ≥60 years of age (112). Of note, in the ACCORD trial the risk of fractures in the women treated with rosiglitazone decreased after discontinuing rosiglitazone therapy (113).
In mice, TZDs suppress bone formation and increase bone resorption resulting in decreased bone mass (85). Additionally, TZD administration in mice results in the massive accumulation of adipocytes in the bone marrow cavity (85). In a meta-analysis of 14 trials with 1,734 participants, treatment with TZDs for 3 to 24 months decreased bone mineral density measured by DEXA at the lumbar spine (difference -1.1%; p < 0.0001), total hip (-1.0%; p < 0.0001) and forearm (-0.9%; p = 0.007) (112). In five studies TZD therapy was discontinued and after 24-52 weeks there was no increase in bone mineral density indicating no restoration of bone mineral density with cessation of TZD treatment (112). In an observation study each year of TZD use was associated with greater bone loss at the whole body (additional loss of -0.61% per year), lumbar spine (-1.23% per year), and trochanter (-0.65% per year) in women, but not men (114).The effect of TZD treatment on bone turnover markers varied considerably between individual studies (112). This reduction in bone mass induced by TZD treatment could contribute to the increase in fractures but it is possible that changes in the microarchitecture of bone also plays a role.
BLADDER CANCER
In preclinical studies pioglitazone administration increased bladder cancer in male rats but not in female rats or in mice, dogs, or monkeys (115). In the PROactive study there was a nonsignificant increase in the number of patients who developed bladder cancer (16 vs 6, p = 0.069) (82). In a number of instances, the development of bladder could not plausibly be related to treatment due to the temporal sequence of drug exposure and cancer diagnosis. After eliminating these patients there were six patients with bladder cancer in the pioglitazone group and three patients in the placebo group (82). After 10 years of follow-up, bladder cancer was reported in 0.8% of patients (n = 14) in the pioglitazone versus 1.2% (n = 21) in the placebo group (relative risk 0.65) during the follow-up period (116). In the IRIS study bladder cancer occurred in 12 patients in the pioglitazone group and in 8 in the placebo group (P=0.37) (83). Thus, in large randomized trials the data do not definitively support that pioglitazone significantly increases the risk of bladder cancer. The short duration of the randomized studies and infrequent occurrence of bladder cancer make interpretation of these studies difficult.
Because of the preclinical data the FDA requested that the manufacturer of pioglitazone initiate a prospective study to examine the relationship between pioglitazone and bladder cancer. This 10-year study of 193,099 persons did not find any statistically significant association between pioglitazone treatment and bladder cancer (117). Additionally, in a multinational cohort of 1.01 million patients with T2DM there was no evidence for any association between cumulative exposure to pioglitazone and bladder cancer in men or women after adjustment for age, calendar year, diabetes duration, smoking and any ever use of pioglitazone (118). Similarly, no association was observed between rosiglitazone and bladder cancer in men or women (118). In a careful review of 23 epidemiological studies Davidson concluded that there was little evidence that pioglitazone increased the risk of bladder cancer (115). The FDA still warns about the possibility of bladder cancer with pioglitazone use and recommends that pioglitazone not be used in diabetic patients with active bladder cancer or history of bladder cancer (package insert).
MACULA EDEMA
Macular edema has been reported in patients taking TZDs (119,120). Patients may present with blurred vision or decreased visual acuity or be diagnosed on routine ophthalmologic examination. Most patients had peripheral edema at the time macular edema was diagnosed (120). Some patients had improvement in their macular edema after discontinuation of the TZD (120).
OVULATION AND PREGNANCY
As discussed above in the polycystic ovary section, treatment of premenopausal women with PCOS may induce ovulation and thereby result in unplanned pregnancies. In premenopausal anovulatory women started on a TZD one needs to discuss the need for contraception.
Contraindications and Drug Interactions
TZDs are contraindicated in patients with NYHA Class III or IV heart failure. Pioglitazone should not be used in diabetic patients with active bladder cancer or history of bladder cancer.
Strong CYP2C8 inhibitors (e.g., gemfibrozil) increase pioglitazone and rosiglitazone concentrations and one should limit pioglitazone dose to 15 mg daily (package insert).
Summary
TZDs are effective drugs in improving glycemic control and have significant benefits on disorders that occur commonly in patients with T2DM (cardiovascular disease, NAFLD/NASH, PCOS). Unfortunately, TZDs also have serious side effects, such as CHF, osteoporosis, and weight gain, that limit their use. Clinicians need to balance the advantages and disadvantages of TZDs for the individual patient.
|
Table 12. The Advantages and Disadvantages of Thiazolidinediones
|
|
Advantages
|
Disadvantages
|
|
Once a day administration
|
Edema
|
|
Reduces CVD (pioglitazone)
|
CHF
|
|
Durable Effect
|
Weight gain
|
|
Reduces NASH
|
Osteoporosis
|
|
No hypoglycemia
|
Bladder cancer (pioglitazone)?
|
|
Relatively inexpensive
|
Macula edema?
|
|
No dose adjustment for renal disease
|
Small increase in LDLc
|
|
Increase HDL-C and decrease triglycerides
|
|
ALPHA-GLUCOSIDASE INHIBITORS
Introduction
Acarbose (Precose, Glucobay), miglitol (Glycet) and voglibose (Basen, Voglib) are members of the α-glucosidase inhibitor class of oral anti-hyperglycemic compounds that were introduced in the 1990s (16).
Administration
The recommended starting dosage of acarbose and miglitol is 25 mg given orally three times daily at the start of each meal. The dose of acarbose and miglitol can be adjusted at 4 to 8-week intervals based on one-hour postprandial glucose or A1c levels, and on tolerance. The dosage can be increased from 25 mg tid with meals to 50 mg tid with meals. The maximum dose is 100 mg tid with meals. Note that the dose can be varied based on the amount of carbohydrate in the meal. In some patients one can initiate therapy once a day with the largest meal.
Mechanism of Action
Alpha-glucosidase inhibitors are competitive, reversible inhibitors of pancreatic α-amylase and membrane-bound intestinal α-glucosidase hydrolase enzymes (16,121). Inhibiting these enzymes prevents the metabolism of disaccharides and oligosaccharides into monosaccharides delaying carbohydrate digestion and absorption (16,121). Carbohydrate absorption occurs more distally in the intestine reducing the postprandial increase in glucose and lowering prandial insulin levels (16,121). Acarbose and miglitol have minimal inhibitory activity against lactase and consequently will not prevent the increase in plasma glucose following the ingestion of milk or cause lactose intolerance (package insert). In addition to effecting carbohydrate absorption, alpha-glucosidase inhibitors increase postprandial GLP-1 secretion and reduce glucose-dependent insulinotropic polypeptide (GIP) secretion (16).
Glycemic Efficacy
The typical decrease in A1c levels is relatively modest with alpha-glucosidase inhibitors (0.5-1.0%) (37,121,122). The decrease in A1c is predominantly due to decreases in post meal glucose levels and alpha-glucosidase inhibitors have only modest effects on fasting glucose levels (16,121,122). Alpha-glucosidase inhibitors can be combined with other hypoglycemic drugs with additive effects and are particularly useful to lower postprandial glucose levels (37,121). Alpha-glucosidase inhibitors are most effective in patients who ingest a high carbohydrate diet and for this reason have been widely used and very effective in Asian populations (16).
These drugs do not cause weight gain and hypoglycemia is uncommon (16,37,122). If a patient experiences hypoglycemia while taking an alpha-glucosidase inhibitor in combination with insulin or sulfonylureas the patient should be instructed to use glucose (gel, tablets, etc.) as alpha-glucosidase inhibitors will prevent the breakdown of sucrose and delay glucose absorption resulting in a failure to quickly correct hypoglycemia. Severe hypoglycemia may require intravenous glucose or intramuscular glucagon administration.
Other Effects
CARDIOVASCULAR DISEASE
In the STOP-NIDDM trial 1,429 subjects with impaired glucose tolerance were randomized to placebo vs. acarbose and followed for 3.3 years (123). In the acarbose group a 49% relative risk reduction in the development of cardiovascular events (hazard ratio 0.51; P =0.03) was observed. Among cardiovascular events, the major reduction was in the risk of myocardial infarction (HR 0.09; P =0.02). In a smaller trial, 135 patients hospitalized for the acute coronary syndrome who were newly diagnosed with IGT were randomly assigned to acarbose or placebo (124). During a mean follow-up of 2.3 years the risk of recurrent major adverse cardiovascular event was decreased significantly in the acarbose group compared with that in control group (26.7% versus 46.9%, P < 0.05).
Despite these favorable observations a large trial failed to demonstrate a beneficial effect of acarbose in Chinese patients with impaired glucose tolerance (ACE trial) (125). In a randomized trial acarbose vs. placebo was compared in 6,522 patients with coronary heart disease and impaired glucose tolerance. The primary outcome was cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, hospital admission for unstable angina, and hospital admission for heart failure and patients were followed up for a median of 5 years. The primary outcome was similar in the acarbose and placebo groups (hazard ratio 0.98; p=0.73). No significant differences were seen for death from any cause, cardiovascular death, fatal or non-fatal myocardial infarction, fatal or non-fatal stroke, hospital admission for unstable angina, hospital admission for heart failure, or impaired renal function.
Thus, whether acarbose favorably affects cardiovascular disease in patients at high risk for developing diabetes is uncertain. Moreover, the effect of acarbose on cardiovascular disease in patients with diabetes is unknown.
WEIGHT
Acarbose is may result in a very small decrease in weight (0.4kg) (126).
Side Effects
Gastrointestinal side effects of alpha-glucosidase inhibitors include flatulence, abdominal discomfort, and diarrhea and are very commonly encountered (16,37,122). These side effects can lead to the inability to tolerate these drugs. A high carbohydrate diet may worsen the GI adverse effects. Over time the GI symptoms tend to decrease as the intestines adapt (121). GI side effects are due to the mechanism of action of alpha-glucosidase inhibitors (121). The inhibition of carbohydrate digestion in the small intestine leads to the delivery of undigested carbohydrates to the large intestine where microorganisms metabolize them into short-chain fatty acids, methane, carbon dioxide, and hydrogen, that can cause abdominal discomfort, increased flatulence, and diarrhea (121).
Acarbose, particularly at doses in excess of 50 mg tid, may give rise to elevations of serum transaminases and, in rare instances, hyperbilirubinemia. It is recommended that serum transaminase levels be checked every 3 months during the first year of treatment with acarbose and periodically thereafter. If elevated transaminases are observed, a reduction in dosage or withdrawal of therapy may be indicated, particularly if the elevations persist (package insert).
Contraindications and Drug Interactions
Acarbose and miglitol are contraindicated in patients with inflammatory bowel disease, colonic ulceration, intestinal obstruction or those predisposed to intestinal obstruction, patients with chronic intestinal disease, or conditions that will be worsened by the increased gas formation in the intestine (37) (package insert). Acarbose is contraindicated in patients with cirrhosis (package insert).
Acarbose and miglitol should not be used in patients with a creatinine > 2 mg/dl (package insert).
Summary
Alpha-glucosidase inhibitors are excellent drugs for lowering postprandial glucose levels. Unfortunately, because of their GI side effects many patients are unable to tolerate these drugs. Additionally, the need for three times a day administration makes it difficult for patients to comply with these drugs.
|
Table 13. Advantages and Disadvantages of Alpha-Glucosidase Inhibitors
|
|
Advantages
|
Disadvantages
|
|
No hypoglycemia
|
GI side effects
|
|
Weight neutral
|
Frequent dosing schedule
|
|
Decreases postprandial glucose
|
Avoid if renal disease (creatinine> 2mg/dL
|
|
Relatively inexpensive
|
|
SODIUM-GLUCOSE TRANSPORT PROTEIN 2 (SGLT2) INHIBITORS
Introduction
There are currently four SGLT2 inhibitors available (Canagliflozin/ Invokana; Dapagliflozin/ Farxiga; Empagliflozin/Jardiance; Ertugliflozin/ Stelgatro) (127). These drugs are very similar and there are only a few differences between these agents.
Administration
The recommended starting dose of canagliflozin is 100 mg once daily, taken before the first meal of the day. In patients tolerating canagliflozin 100 mg once daily who have an eGFR of 60 mL/min/1.73 m2 or greater and require additional glycemic control, the dose can be increased to 300 mg once daily.
The recommended starting dose of dapagliflozin is 5 mg once daily, taken in the morning, with or without food. In patients tolerating dapagliflozin 5 mg once daily who require additional glycemic control, the dose can be increased to 10 mg once daily.
The recommended starting dose of empagliflozin is 10 mg once daily in the morning, taken with or without food. In patients tolerating empagliflozin, the dose may be increased to 25 mg.
The recommended starting dose of ertugliflozin is 5 mg once daily, taken in the morning, with or without food. In patients tolerating ertugliflozin 5 mg once daily who require additional glycemic control, the dose can be increased to 15 mg once daily.
Before initiating SGLT2 inhibitor therapy one should assess renal function and volume status. The dose of SGLT2 inhibitors may need to be adjusted based on renal function (see below).
Mechanism of Action
SGLT2 is a low-affinity, high-capacity glucose transporter in the proximal tubules of the kidneys, which is responsible for the reabsorption of the majority of the filtered glucose (approximately 90%) entering the tubules (16,128). SGLT1, which is predominantly expressed in the intestines is also expressed in the kidneys, is a high-affinity, low-capacity glucose transporter in the proximal tubules, which makes a minor contribution to the reabsorption of filtered glucose (approximately 10%) (16,128). SGLT 1 and 2 transporters are capable of reabsorbing virtually all the filtered glucose when blood glucose levels are less than approximately 180mg/dL. When blood glucose levels are greater than approximately 180mg/dL, glucose begins to appear in the urine (i.e., glycosuria). The higher the blood glucose level the greater the quantity of glucose in the urine. Patients with T2DM express a greater number of SGLT2 transporters in the proximal tubule than do healthy individuals and hence glucose reabsorption from the glomerular filtrate is increased in patients with diabetes and glycosuria occurs at a higher blood glucose level (typically approximately 220mg/dl (129).
Inhibition of SGLT2 by drugs results in glycosuria and can lead to the excretion of 60–90 grams of glucose in the urine per day (Figure 8) (16). The amount of glucose excreted in the urine can vary considerably depending on renal function and the degree of hyperglycemia (16). Decreased renal function results in a decrease in filtered glucose and less glucose in the urine while high blood glucose levels increase filtered glucose and increases the loss of glucose in the urine (16). The ability of the inhibition of SGLT2 to lower blood glucose levels is not dependent on insulin action and hence is not affected by insulin levels or insulin resistance (16). As will be discussed below many of the non-glucose lowering benefits and side effects of SGLT2 inhibitors can be explained by the increase in glucose excretion in the urine. It should be recognized that glycosuria results in an osmotic diuresis. Additionally, because the SGLT2 transporters also facilitate the reabsorption of sodium from the filtrate there is also the loss of sodium in the urine.
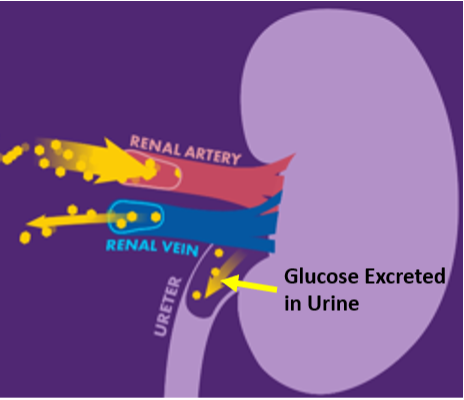
Figure 8. Effect of SGLT2 Inhibitors on the Kidney
Glycemic Efficacy
A meta-analysis of 66 randomized trials found that SGLT2 inhibitors decreased A1c levels by 0.4 to 1.1% (130). In comparison to other hypoglycemic drugs, it was found that SGLT‐2 inhibitors showed a greater efficacy than DPP‐4 inhibitors and similar or slightly less efficacy compared to metformin and TZDs (12,130). Sulfonylureas appeared to be superior to SGLT‐2 inhibitors at 12 weeks, but at 24- and 52-weeks efficacy was similar or slightly lower (12,130). However, SGLT‐2 inhibitors produced a greater reduction in HbA1c than sulfonylureas at 104 weeks perhaps due to the lack of durability of sulfonylurea therapy discussed earlier (130). The A1c lowering ability of the different SGLT2 inhibitors is similar but A1c is reduced to a slightly greater extent by high-dose canagliflozin, which is probably a result of its additional action of inhibiting SGLT1 in the intestine decreasing dietary glucose absorption (127,128,130). SGLT2 inhibitors when used as an add-on therapy to metformin, insulin, thiazolidinediones, DPP-4 inhibitors, GLP-1 receptor agonists, sulfonylureas, or metformin ± DPP-4 inhibitor were similarly effective in reducing A1c levels as when used in monotherapy (16,128). The efficacy of SGLT2 inhibitors is dependent on renal function and as renal function decreases the ability of these drugs to lower A1c levels diminishes (16,128). SGLT2 inhibitors lower both fasting and postprandial glucose levels (128). In monotherapy SGLT2 inhibitors have a low risk of causing hypoglycemia but in combinations with insulin or sulfonylureas may potentiate the development of hypoglycemia (16). In patients in good glycemic control, one often decreases the insulin or sulfonylurea dose when initiating therapy with an SGLT2 inhibitor.
Other Effects
WEIGHT
SGLT2 inhibitors lead to weight loss (16,128). In general patient’s lose approximately 1- 3 kg on these drugs (16,127,128). SGLT2 inhibitor-induced weight loss results primarily from a decrease in fat mass, including reductions in visceral and subcutaneous adipose tissue (128). The weight loss is due to the loss of glucose in the urine, which represents the loss of calories (128,131). The excretion of 50 grams of glucose in the urine is equivalent to the loss of 225 calories (50-grams X 4.5 calories per gram of glucose). However, the amount of glucose lost in the urine should result in a greater weight loss than is typically observed and a compensatory increase in food intake blunts the weight loss (131). There are likely to be other homeostatic mechanisms that also play a role in limiting weight loss with SGLT2 inhibitors.
GLUCOSE MONITORING
Monitoring glycemic control with 1,5-AG assay is not accurate as measurements of 1,5-AG are unreliable in patients taking SGLT2 inhibitors.
BLOOD PRESSURE
SGLT2 inhibitors decrease systolic BP by approximately 3-6 mmHg and diastolic BP by approximately 2-3 mmHg (16,128). Patients with poorly controlled BP at baseline experience the largest reduction in BP (127). SGLT2 inhibitors lower BP by promoting an osmotic diuresis and decreasing intravascular volume (128). Weight loss may also contribute to the decrease in BP.
LIPID LEVELS
SGLT2 inhibitors cause a small increase in LDL and HDL cholesterol levels. In the EMPA-REG outcome study, described in detail below, LDL cholesterol levels were increased by 2-4 mg/dL and HDL cholesterol by 2-3 mg/dL in the group treated with empagliflozin (132). Similarly, in the CANVAS outcome study, discussed in detail below, LDL cholesterol and HDL cholesterol were also marginally increased in the canagliflozin treated group (LDL cholesterol 4-5 mg/dL and HDL cholesterol 2-3 mg/dL) (133). In a meta-analysis of 43 randomized trials with 22,528 patient’s triglyceride levels were decreased by 2 mg/dL (134). In a meta-analysis of 48 randomized controlled trials SGLT2 inhibitors significantly increased LDL-C (3.8mg/dl, p < 0.00001), HDL-C (2.3mg/dl, p < 0.00001), and decreased triglyceride levels (8.8mg/dl, p < 0.00001) (135). It is unlikely that these small changes in LDL-C, HDL-C, and triglyceride levels are of clinical significance. The mechanism for these increases in LDL and HDL cholesterol is unknown but could be due to a decrease in plasma volume. The decrease in triglycerides might be secondary to weight loss.
URIC ACID
SGLT2 inhibitors lower blood uric acid levels (136). This decrease is due to an increase in uric acid excretion by the kidneys. In an observational study 47,905 individuals receiving an SGLT2 inhibitor and 183,303 receiving a DPP4 inhibitor it was observed that the incidence of gout was 20.26 per 1000 patient-years for SGLT2 inhibitor users and 24.30 per 1000 patient-years for DPP4 inhibitor users (137).
CARDIOVASCULAR
There have been several large randomized studies of the effect of SGLT2 inhibitors on cardiovascular events published (others are in progress).
EMPA-REG Outcome Trial
In this study, 7,020 patients with established cardiovascular disease and T2DM were randomly assigned to receive 10 mg or 25 mg of empagliflozin or placebo once daily and were followed for 3.1 years (132). In the combined empagliflozin treated groups there was a statistically significant 14% reduction in the primary outcome (death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke). As compared with placebo, empagliflozin treatment did not result in a significant difference in the occurrence of non-fatal myocardial infarction or strokes. However, empagliflozin resulted in a significantly lower risk of death from cardiovascular causes (hazard ratio, 0.62), death from any cause (hazard ratio, 0.68), and hospitalization for heart failure (hazard ratio, 0.65). The beneficial effects of empagliflozin were noted to occur very rapidly and the beneficial effects on heart failure appeared to be the dominant effect compared to effects on atherosclerotic events. Decreases in cardiovascular outcomes and mortality with empagliflozin occurred across the range of cardiovascular risk (138). Additionally, the reduction in hospitalizations for heart failure and cardiovascular death were observed both in patients with and without heart failure at baseline (139).
CANVAS Trial
The effects of placebo vs. canagliflozin 100mg or 300mg per day were determined in two combined trials involving a total of 10,142 participants with T2DM and high cardiovascular risk (approximately 70% of patients had established cardiovascular disease) (133). The primary outcome was a composite of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke and the mean follow-up was 188 weeks. The primary outcome was reduced in the canagliflozin group (hazard ratio, 0.86; P=0.02). The effect of canagliflozin on the primary outcome was similar in people with chronic kidney disease and those with preserved kidney function (140). Death from any cause (hazard ratio 0.87; 95% CI 0.74-1.01) and death from cardiovascular disease (hazard ratio 0.87; 95% CI 0.72-1.06) were reduced but were not statistically significant. Similarly, canagliflozin treatment did not result in a significant difference in non-fatal strokes or non-fatal myocardial infarctions (hazard ratio 0.90 for stroke and 0.85 for myocardial infarction). As seen with empagliflozin, hospitalization for heart failure was markedly reduced (hazard ratio 0.67; 95% CI 0.52-0.87) and this beneficial effect occurred rapidly.
CREDENCE Trial
In a second canagliflozin trial that focused on patients with kidney disease, a decrease in cardiovascular events was also observed (141). In this double-blind trial 4,401 patients with chronic kidney disease and T2DM were randomized to canagliflozin 100mg per day or placebo and followed for a median of 2.62 years. All the patients had an eGFR of 30 to <90 ml per minute per 1.73 m2 and albuminuria (ratio of albumin [mg] to creatinine [g], >300 to 5000). In this trial hospitalization for heart failure was reduced by 39%. The relative benefits of canagliflozin for cardiovascular outcomes was similar in individuals across the spectrum of eGFR levels (142)
DECLARE–TIMI 58 Trial
The effect of dapagliflozin on cardiovascular events has been reported (143). 17,160 patients with T2DM, including 10,186 without atherosclerotic cardiovascular disease, were randomized to dapagliflozin 10mg per day or placebo and followed for a median of 4.2 years. The primary outcome was a composite of major adverse cardiovascular events (MACE), defined as cardiovascular death, myocardial infarction, or ischemic stroke. The primary efficacy outcomes were MACE and a composite of cardiovascular death or hospitalization for heart failure. Dapagliflozin did not result in a lower rate of major adverse cardiovascular events (8.8% in the dapagliflozin group and 9.4% in the placebo group; hazard ratio, 0.93; P=0.17) but did result in a lower rate of cardiovascular death or hospitalization for heart failure (4.9% vs. 5.8%; hazard ratio, 0.83; P=0.005), which reflected a lower rate of hospitalization for heart failure (hazard ratio, 0.73; 95% CI, 0.61 to 0.88). Interestingly, in the patients with a history of a previous MI dapagliflozin reduced the risk of a MACE (HR 0.84; P=0.039), whereas there was no effect in patients without a previous MI (144). Dapagliflozin reduced the risk of heart failure in patients with and without a history of heart failure but the benefit was greater in patients with a history of heart failure (with heart failure HR 0.62; 95% CI, 0.45-0.86; without heart failure HR 0.88; 95% CI, 0.74-1.03) (145). Dapagliflozin also reduced the risk of heart failure in patients without a history of atherosclerotic cardiovascular disease (146). In addition, dapagliflozin reduced the risk of atrial fibrillation and atrial flutter by 19% (HR, 0.81; P=0.009) (147).
VERTIS CV
Patients with atherosclerotic cardiovascular disease and T2DM were randomized to ertugliflozin 5mg (n=2752), 15mg (2747), or placebo (n=2747) and the primary composite outcome of cardiovascular death and non-fatal MI or stroke was determined after a mean duration of follow-up of 3.5 years (148). This trial did not demonstrate a significant difference in the primary endpoint (MACE) nor any components of the primary endpoint. However, heart failure hospitalizations were significantly reduced by 30% in the patients treated with ertugliflozin (HR 0.70; CI 0.54–0.90). The benefits on heart failure were observed in both patients with a history of heart failure (decreased 37%) and patients without a history of heart failure (decreased 21%) (149).
DAPA HF Trial
In this trial 4,744 patients with New York Heart Association class II, III, or IV heart failure and an ejection fraction of 40% or less were randomized to receive either dapagliflozin 10 mg once daily or placebo for a median of 18.2 months (150). The primary outcome was a composite of worsening heart failure (hospitalization or an urgent visit resulting in intravenous therapy for heart failure) or cardiovascular death. Of note only approximately 45% of the patients had type 2 diabetes. Treatment with dapagliflozin reduced the primary outcome (HR 0.74; 95% CI, 0.65 to 0.85; P<0.001), heart failure (HR 0.70; 95% CI, 0.59 to 0.83), and death from cardiovascular disease (HR 0.82; 95% CI, 0.69 to 0.98). Symptoms of heart failure were also improved with dapagliflozin treatment. Additionally, dapagliflozin reduced the risk of any serious ventricular arrhythmia, cardiac arrest, or sudden death (151). The benefits of dapagliflozin were similar in patients with diabetes and the non-diabetic patients (152). This study demonstrates that an SGLT2 inhibitor is beneficial in patients with pre-existing heart failure and this occurs in both patients with and without diabetes.
EMPEROR-Reduced Trial
In this trial 3,730 patients with class II, III, or IV heart failure and an ejection fraction of 40% or less were randomized to empagliflozin 10 mg once daily or placebo for a median of 16 months (153). The primary outcome was a composite of cardiovascular death or hospitalization for heart failure. Approximately 50% of the patients had type 2 diabetes. Treatment with empagliflozin reduced the primary outcome (HR 0.75; 95% CI, 0.65 to 0.86; P<0.001) and hospitalization for heart failure (HR 0.69; 95% CI, 0.59 to 0.81) but did not reduce cardiovascular death (HR 0.92; 95% CI, 0.75 to 1.12). The beneficial effects were observed in patients with and without diabetes. This study is concordant with the results observed in the DAPA HF trial and demonstrates that SGLT2 inhibitors are beneficial in patients with pre-existing heart failure and this occurs in both patients with and without diabetes.
DAPA CKD Trial
This trial randomly assigned 4,304 participants with an eGFR of 25 to 75 ml/min/1.73 m2 of body-surface area and a urinary albumin-to-creatinine ratio 200 to 5000 mg/g to receive dapagliflozin 10 mg once daily or placebo for a median of 2.4 years (154). Approximately 67% of the patients had diabetes. The composite of death from cardiovascular causes or hospitalization for heart failure was decreased in the dapagliflozin group (HR 0.71 95% CI, 0.55–0.92).
EMPEROR-Preserved Trial
This trial randomized 5,988 patients with heart failure with an ejection fraction of >40% to double-blind treatment with placebo or empagliflozin 10 mg once daily (155). Empagliflozin decreased the combined risk of cardiovascular death, hospitalization for heart failure, or an emergency or urgent heart failure visit by 23% (HR 0.77; P<0.0001). Moreover, this benefit occurred rapidly reaching statistical significance at 18 days after randomization. The benefit of empagliflozin was similar in patients with an ejection fraction of >40% to <50% and 50% to <60%, but was attenuated at higher ejection fractions. These results indicate that SGLT2 inhibitors are beneficial even in patients with a preserved ejection fraction.
Summary
Thus, nine randomized trials of SGLT2 inhibitors demonstrated a robust decrease in heart failure (table 14) with SGLT2 inhibitor therapy without a consistent strong effect on myocardial infarctions or strokes. In a meta-analysis of eight of these trials (not including Emperor Preserved) with 59,747 patients it was observed that SGLT2 inhibitors reduced the risk of all-cause mortality (HR 0.84; 95% CI [0.78-0.91]), cardiovascular mortality (HR 0.84; 95% CI [0.76-0.93]) hospitalization for heart failure (HR 0.69; 95% CI [0.64-0.74]), and myocardial infarction (HR 0.91; 95% CI [0.84-0.99]), but there was no significant effect on the risk of stroke (HR 0.98; 95% CI [0.86-1.11]) (156). The reduction in heart failure was seen in patients with and without diabetes, patients with renal disease, and patients with and without a history of heart failure. The Emperor Preserved trial demonstrated that patients with a preserved ejection fraction also benefit from treatment with a SGLT2 inhibitor. Finally, the EMPULSE trial demonstrated that starting empagliflozin during the hospitalization for heart failure was beneficial (157). For additional information see the section on drugs that inhibit both SGLT1 and 2.
|
Table 14. Summary of Effect of SGLT2 Inhibitors on Heart Failure
|
|
|
Number
|
Prior Heart Failure
|
Mean Follow-up (years)
|
Hazard Ratio* (95% CI)
|
P value
|
|
EMPA-REG
Empagliflozin
|
7,020
|
10.1%
|
3.1
|
0.65
(0.05-0.85)
|
0.002
|
|
CANVAS
Canagliflozin
|
10,142
|
14.4%
|
3.6
|
0.67
(0.52-0.87)
|
--
|
|
DECLARE-TIMI 58
Dapagliflozin
|
17,160
|
10.0%
|
4.2
|
0.73
(0.61-0.88)
|
0.0007
|
|
VERTIS-CV
Ertugliflozin
|
8,246
|
23.7%
|
3.0
|
0.70
(0.54-0.90)
|
0.006
|
|
CREDENCE
Canagliflozin
|
4,401
|
14.8%
|
2.6
|
0.61
(0.47-0.80
|
0.001
|
|
DAPA-HF
Dapagliflozin
|
4,774
|
100%
|
1.5
|
0.70
(0.59-0.83)
|
0.001
|
|
EMPEROR
Empagliflozin
|
3,730
|
100%
|
1.3
|
0.69
(0.59-0.81)
|
<0.001
|
|
EMPEROR Preserved
|
5,988
|
100%
|
2.2
|
0.73
(0.61 to 0.88)
|
<0.001
|
|
DAPA-CKD
|
4,304
|
11%
|
2.4
|
0.71**
(0.55–0.92)
|
<0.009
|
*Hospitalization for Heart Failure
** Hospitalization for Heart Failure and death from cardiovascular disease
The mechanisms accounting for the beneficial effects of SGLT2 inhibitors on heart failure are uncertain (158). Glycemic control was better in the SGLT2 inhibitor treated patients but it is doubtful that this modest decrease in glucose could account for the observed results (additionally benefit in non-diabetics makes a glucose effect very unlikely). SGLT2 inhibitor treatment was associated with small reductions in weight, waist circumference, uric acid level, and systolic and diastolic blood pressure, with no increase in heart rate and small increases in both LDL and HDL cholesterol. Whether these changes played a role in reducing events remains to be determined but it is unlikely that these play a major role as other treatments that effect these factors do not markedly diminish the risk of heart failure events. It is possible that hemodynamic changes secondary to the osmotic diuresis induced by SGLT2 inhibitors contributed to the beneficial effects. In an analysis of the EMPA-REG OUTCOME trial, the change in hematocrit (~3% increase), corresponding to ~7% reduction in plasma volume, accounted for approximately 50% of the benefit of the drug on cardiovascular death (159). Additionally, SGLT2 inhibitors increase free fatty acid levels and glucagon secretion, which promotes the production of ketone bodies such as beta-hydroxybutyrate that are utilized by the heart for energy production (160). It is possible that this alternative source of energy could be protective for heart function. Finally, there may be direct effects of SGLT2 inhibition on myocardial and renal metabolism (158,161,162). Further studies are required to better elucidate the mechanism of the beneficial effects of SGLT2 inhibitors on heart failure.
RENAL DISEASE
The large randomized SGLT2 inhibitor cardiovascular outcome trials described above also examined the effect of these drugs on renal disease.
EMPA-REG Outcome Trial
The effect of empagliflozin on renal outcomes was studied in 4,124 patients with T2DM who were randomized to empagliflozin (10 mg or 25 mg) or placebo (163). The prespecified outcomes were progression to macroalbuminuria, doubling of the serum creatinine level, initiation of renal-replacement therapy, or death from renal disease and incident albuminuria. Worsening nephropathy occurred in 12.7% of patients in the empagliflozin group and in 18.8% of patients in the placebo group, a relative risk reduction of 39% (P<0.001). Progression to macroalbuminuria was reduced 38%, doubling of serum creatinine by 44%, and initiation of renal replacement therapy by 55% (all statistically significant). The renal benefit was seen regardless of baseline eGFR, occurring in individuals with an eGFR as low as 30 mL/min/1.73 m2. While empagliflozin caused an initial decrease in eGFR over the long term eGFR decreased in the placebo group at a more rapid rate than the empagliflozin group. Additionally, patients treated with empagliflozin were more likely to convert from microalbuminuria to normoalbuminuria (hazard ratio [HR] 1.43; p<0·0001) or from macroalbuminuria to microalbuminuria or normoalbuminuria (HR 1.82; p<0·0001), and were less likely to experience a sustained deterioration from normoalbuminuria to microalbuminuria or macroalbuminuria (HR 0·84; p=0·0077) (164).
CANVAS Trial
Similar to the results seen with empagliflozin, canagliflozin has also been shown to decrease renal disease. 10,142 participants with T2DM and high cardiovascular risk were randomly assigned to receive canagliflozin or placebo and were followed for a mean of 188.2 weeks (133). Progression of albuminuria occurred less frequently in the canagliflozin group (hazard ratio of 0.73; 95% CI, 0.67 to 0.79). In addition, regression of albuminuria also occurred more frequently in the canagliflozin group (hazard ratio, 1.70; 95% CI, 1.51 to 1.91). Most importantly, the composite outcome of sustained 40% reduction in eGFR, the need for renal-replacement therapy, or death from renal causes occurred less frequently in the canagliflozin group (hazard ratio of 0.60; 95% CI, 0.47 to 0.77). Annual eGFR decline was slower (slope difference between groups 1.2 mL/min/1.73 m2 per year, 95% CI 1.0-1.4) and mean urinary albumin creatinine ratio was 18% lower (95% CI 16-20) in participants treated with canagliflozin than in those treated with placebo (165). The benefits of canagliflozin on renal disease occurred across a wide spectrum of eGFR ranging from 30-45 to ≥90 mL/min/1.73 m2 and in patients with moderate and severe albuminuria (140,166).
CREDENCE Trial
The CREDENCE Trial focused on patients with renal disease. In a double-blind trial 4,401 patients with T2DM and chronic kidney disease were randomized to canagliflozin or placebo and followed for a median of 2.62 years (141). All the patients had an eGFR of 30 to <90 mL/min/1.73 m2 and albuminuria (ratio of albumin [mg] to creatinine [g], >300 to 5000) and were treated with renin-angiotensin system blockade. The primary outcome was a composite of end-stage kidney disease (dialysis, transplantation, or a sustained estimated GFR of <15 mL/min/1.73 m2), a doubling of the serum creatinine level, or death from renal or cardiovascular causes. The primary outcome was 30% lower in the canagliflozin group (hazard ratio, 0.70; P = 0.00001). The relative risk of the renal-specific composite of end-stage kidney disease, a doubling of the creatinine level, or death from renal causes was 34% lower (hazard ratio, 0.66; P<0.001), and the relative risk of end-stage kidney disease was 32% lower (hazard ratio, 0.68; P = 0.002). Benefits were seen regardless of baseline eGFR.
DECLARE–TIMI 58 Trial
In this trial of 17,160 participants a secondary outcome was a renal composite outcome defined as a sustained decrease of 40% or more in eGFR to less than 60 mL/min/1.73 m2, new end-stage renal disease, or death from renal or cardiovascular causes (143). As seen in the other SGLT2 inhibitor studies there was a decrease in the development of renal disease with the incidence of the renal outcome 4.3% in the dapagliflozin group vs. 5.6% in the placebo group (hazard ratio, 0.76; 95% CI, 0.67 to 0.87). Excluding death from cardiovascular causes as part of the composite endpoint, the reduction in renal events was even more impressive (HR 0.53 p<0.0001) (167). The risk of end-stage renal disease or renal death was lower in the dapagliflozin group than in the placebo group (HR 0.41; p=0.012) (167).
VERTIS CV Trial
In VERTIS CV trial the renal composite end point of renal death, dialysis/transplant, or doubling of serum creatinine was reduced but not statistically significant in the ertugliflozin treated group (HR 0.81; CI 0.63–1.04) (148).
DAPA-HF Trial
In this trial 4,744 patients with New York Heart Association class II, III, or IV heart failure and an ejection fraction of 40% or less were randomized to receive either dapagliflozin 10 mg once daily) or placebo for a median of 18.2 months (150). The renal outcome was a composite outcome of a reduction of 50% or more in the estimated GFR sustained for at least 28 days, end-stage renal disease, or death from renal causes. End-stage renal disease was defined as an eGFR of less than 15 ml/min/1.73 m2, long-term dialysis, or kidney transplantation. There was a trend towards benefit with dapagliflozin treatment that was not statistically significant due to a small number of events (HR 0.71; 95% CI 0.44 to 1.16).
EMPEROR-Reduced Trial
In this trial 3,730 patients with class II, III, or IV heart failure and an ejection fraction of 40% or less were randomized to empagliflozin 10 mg once daily or placebo for a median of 16 months (153). The annual rate of decline in the eGFR was decreased in the empagliflozin group compared to the placebo group (-0.55 vs. -2.28 ml per minute per 1.73 m2 of body-surface area per year, P<0.001). Additionally, a composite renal outcome (chronic dialysis or renal transplantation or a profound, sustained reduction in the eGFR) was decreased in the empagliflozin group (HR 0.50; 95% CI, 0.32 to 0.77).
DAPA-CKD Trial
In this trial 4,304 individuals with an eGFR) of 25 to 75 ml/minute/m2 and a urinary albumin-to-creatinine ratio of 200 to 5000mg/g were randomized to dapagliflozin 10 mg/day or placebo for a median of 2.4 years (this study was stopped early by the data monitoring board) (154). The primary outcome was a composite of a sustained decline in the estimated GFR of at least 50%, end-stage kidney disease, or death from renal or cardiovascular causes and this was reduced by 39% in the dapagliflozin group (HR 0.61; 95%l CI 0.51 to 0.72; P<0.001; number needed to treat to prevent one primary outcome event, 19). All of the components of this primary outcome were decreased in the dapagliflozin group. A sustained decline in the estimated GFR of at least 50%, end-stage kidney disease, or death from renal causes was reduced by 44% in the dapagliflozin group (HR 0.56; P<0.001). In the subgroup of patients with Stage 4 chronic kidney disease (eGFR< 30) the benefits of dapagliflozin were similar to those described above indicating that even in patients with severe renal disease dapagliflozin is beneficial (168) Finally, the benefits of dapagliflozin were similar in participants with type 2 diabetes (36% decrease) and in those without type 2 diabetes (50% decrease). Thus, similar to the CREDENCE trial, this trial demonstrates that dapagliflozin decreases renal disease progression in patients with pre-existing renal disease. Moreover, this benefit is seen in patients with and without diabetes. Finally, benefit was observed in the dapagliflozin group regardless of the type of kidney disease (diabetic, ischemic, hypertensive, glomerulonephritis, other, or unknown) (169).
Summary
Multiple trials clearly demonstrate that SGLT2 inhibitors have beneficial effects on renal function and decrease the development and progression of renal disease (Table 15). In a meta-analysis of these 8 trials with 59,747 patients there was a robust decrease in the composite end points of renal disease (HR 0.62; 95% CI, 0.56-0.70) (156). The benefits are observed in patients with and without diabetes, with and without renal disease, and also in patients with heart failure. In a smaller meta-analysis this renal disease benefit was seen in patients with and without atherosclerosis (170). These renal benefits are independent of improvement in glycemic control and occurs in patients without diabetes (171).
|
Table 15. Summary of SGLT2 Inhibitors on Renal Disease
|
|
|
Number
|
Mean Follow-up (years)
|
Hazard Ratio* (95% CI)
|
|
EMPA-REG; Empagliflozin
|
7,020
|
3.1
|
0.54 (0.40-0.75
|
|
CANVAS; Canagliflozin
|
10,142
|
3.6
|
0.60 (0.47-0.77)
|
|
DECLARE-TIMI 58; Dapagliflozin
|
17,160
|
4.2
|
0.53 (0.43-0.66)
|
|
VERTIS-CV; Ertugliflozin
|
8,246
|
3.0
|
0.81 (0.63-1.04)
|
|
CREDENCE; Canagliflozin
|
4,401
|
2.6
|
0.66 (0.53-0.81)
|
|
DAPA-HF; Dapagliflozin
|
4,774
|
1.5
|
0.71 (0.44-1.16)
|
|
EMPEROR; Empagliflozin
|
3,730
|
1.3
|
0.52 (0.32-0.77)
|
|
DAPA-CKD; Dapagliflozin
|
4304
|
2.4
|
0.56 (0.45-0.68)
|
*Renal composite outcomes
The mechanism accounting for this effect is unknown but a leading hypothesis is that an increase of sodium chloride in the macula densa due to SGLT2 inhibition triggers a cascade that reduces GFR through constriction of the afferent glomerular arterioles (tubuloglomerular feedback) (128,171). This would reduce glomerular hydrostatic pressure and initially decrease GFR, an effect that is observed with SGLT2 treatment, but in the long run this decrease in GFR protects the kidney from damage resulting in improved kidney function long-term (128).
NONALCOHOLIC FATTY LIVER DISEASE (NAFLD) AND NONALCOHOLIC STEATOHEPATITIS (NASH)
Numerous studies have shown that treatment with SGLT-2 inhibitors decrease liver enzymes (96,172-176). Moreover, studies have shown a decrease in liver fat and liver stiffness (96,172,173,175-177). A study of 5 patients showed an improvement in liver histology after 24 weeks of therapy with canagliflozin (178). Further studies are required to determine whether SGLT-2 inhibitors will result in clinical benefits in patients with NAFLD and NASH.
MORTALITY
A meta-analysis of 21 randomized controlled trials with 70,364 individuals reported that all-cause mortality was decreased by 14% (179). The decrease in all-cause mortality was seen with all of the SGLT2 inhibitors but was not statistically significant with ertugliflozin.
Side Effects
In a meta-analysis of 51 randomized controlled trials involving 24,371 patients it was noted that the frequency of side effects was similar with high dose and low dose SGLT-2 inhibitors (180).
URINARY TRACT INFECTIONS
In some but not all studies an increased risk of urinary tract infections was observed with SGLT2 inhibitors (16,127). In the large randomized cardiovascular outcome trials, an increase in urinary tract infections were not observed (132,133,143). In a meta-analysis of 10 large outcome trials with 71,553 participants the relative risk of urinary tract infection was minimal (RR 1.06, 95% CI 1.00-1.12) (181). In a large meta-analysis of 86 randomized trials with 50,880 patients an increase in urinary tract infections was also not observed (182). The potential increase in the occurrence and severity of urinary tract infections is due to the glycosuria as glucose is an excellent substrate for the growth of micro-organisms.
GENITAL MYCOTIC INFECTIONS
Genital mycotic infections (mainly balanitis and vulvovaginitis) are increased with SGLT2 inhibitor treatment (127). The risk of genital mycotic infections is greater in women than men. In a meta-analysis that included over 2000 patients treated with canagliflozin 100 mg or 300 mg vs. placebo, genital mycotic infections were seen in greater than 10% of women (100mg-10.4%, 300 mg-11.4%, placebo-3.2%) and around 4% of men (100 mg-4.2%, 300 mg-3.7%, placebo- 0.6%) (183). In uncircumcised men the risk of genital mycotic infections is greater than in circumcised men. Genital mycotic infections are the most common side effect seen with SGLT2 inhibitors but fortunately these infections are generally mild and relatively easy to treat (16).
The increase in genital mycotic infections is due to the glycosuria as glucose is an excellent substrate for the growth of Candida.
FOURNIER GANGRENE
Fournier gangrene (FG) is a necrotizing fasciitis of the perineum that is characterized by a rapidly progressive necrotizing infection of the external genitalia, perineum, and perianal region (184). Many of the patients with FG have diabetes (32-66%) (184). FG occurs most commonly in males and is a rare condition with an incidence of 3.3 in 100,000 men aged 50 to 79 years (184). In a recent case series of 59 patients over a 10-year period at a single institution, the incidence was estimated at 32 cases per 100,000 admissions (185). Risk factors included very high A1c (mean 9.6%), obesity, immunocompromised state, and illicit drug use (185). FG is a urologic emergency and requires treatment with broad-spectrum antibiotics and immediate surgical intervention (184).
A recent report described 55 FG cases in patients treated with SGLT2 inhibitors in the last 6 years since they were approved for use in the US (184). In contrast, only 19 cases of FG were reported in 35 years among patients receiving other hypoglycemic drugs. All of the SGLT2 inhibitors were associated with FG except ertugliflozin, which is likely explained by this drug only recently being approved for the treatment of diabetes. However, the authors were unable to assess the incidence of FG or whether SGLT2 inhibitors were causative. A second study compared the occurrence of FG in patients treated with SGLT2 inhibitors (15.0 per 100,000 person-years) vs DPP4 inhibitors (9.7 per 100 000 person-years) in men 65 years and older who have T2DM using large data bases (186).
Early recognition of FG is essential to reduce morbidity and mortality. Typical presentations include systemic symptoms, such as fatigue, fever, and malaise, and local symptoms that include tenderness, erythema, and swelling (184). Pain out of proportion to the clinical findings is highly suggestive of necrotizing fasciitis (184).
HYPOVOLEMIA AND HYPOTENSION
SGLT2 inhibitors induce an osmotic diuresis (127). This effect can result in postural dizziness, orthostatic hypotension, falls, and dehydration, particularly in elderly individuals, patients with kidney disease, patients on either diuretics or medications that interfere with the renin-angiotensin-aldosterone system (e.g., angiotensin-converting-enzyme inhibitors, angiotensin receptor blockers), and patients with low systolic blood pressure (127) (package insert). In a meta-analysis of 10 large outcome studies the risk of volume depletion was modestly increased (RR 1.14, 95% CI 1.06-1.23) (181). Volume status should be determined prior to initiating therapy with an SGLT2 inhibitor.
ACUTE KIDNEY INJURY
SGLT2 inhibitors have been reported to cause acute kidney injury (127). It is likely that volume depletion and hypotension lead to the acute kidney injury (127). In an analysis of two large health care utilization cohorts SGLT2 inhibitors were not associated with an increased risk of acute kidney injury (187). Similarly, in the cardiovascular outcome studies described earlier an increase in acute kidney injury was not observed. In fact, in a meta-analysis of 4 large studies (EMPA-REG, CANVAS, CREDENCE, and DECLARE-TIMI 58) a decrease in acute kidney injury was observed (Risk ratio 0.75; p<0.0001) (188). Similarly, a meta-analysis of 10 studies with 71,553 participants also did not observe an increase in acute kidney injury and in fact observed a decrease (RR 0.84, 95% CI 0.77-0.91) (181). Even in patients over age 75 years of age an increase in acute kidney injury was not observed with SGLT2 treatment (189).
Before initiating SGLT2 inhibitor therapy one should consider factors that may predispose patients to acute kidney injury including hypovolemia, chronic renal insufficiency, congestive heart failure, and concomitant medications (diuretics, ACE inhibitors, ARBs, NSAIDs). Consider temporarily discontinuing SGLT2 inhibitors in any setting of reduced oral intake (such as acute illness or fasting) or fluid losses (such as gastrointestinal illness or excessive heat exposure) (package insert).
DIABETIC KETOACIDOSIS
Diabetic ketoacidosis (DKA) has been observed in patients with T2DM treated with SGLT2 inhibitors but is a rare side effect (16,127). In some instances, the glucose levels are not very elevated despite the patient having DKA (euglycemic DKA) and this can result in a delay in diagnosing DKA (127). SGLT2 inhibitors were associated with approximately twice the risk of diabetic ketoacidosis compared to treatment with DPP-4 inhibitors (190). Additionally, in several of the large cardiovascular studies described above an increase in DKA was observed (CANVAS Trial- canagliflozin 0.6 vs. placebo 0.3 participants with an event per 1000 patient-years; CREDENCE Trial- canagliflozin 2.2 vs. placebo 0.2 per with an event per 1000 patient-years; DECLARE–TIMI 58-dapagliflozin 27 episodes vs placebo 12 episodes; VERTIS trial 0.3% 5mg ertugliflozin, 0.4% 15mg dose, and 0.1% placebo group) (133,141,143,148). In a meta-analysis of 10 studies with 71,553 participants the risk of DKA was increased (RR 2.23, 95% CI 1.36-3.63) (181).
Many of the DKA events occurred in patients with T2DM treated with insulin who had reduced or stopped insulin or experienced an intercurrent illness that could precipitate DKA (16,191). In some instances, the patients were thought to have T2DM but actually had latent autoimmune diabetes of adults (LADA), a form of Type 1 diabetes (16). The hyperglycemia in DKA associated with SGLT2 inhibitors is typically mild because the SGLT2 inhibitors reduce blood glucose levels (16). SGLT2 inhibitors should be temporarily discontinued in clinical situations known to predispose to ketoacidosis (e.g., prolonged fasting due to acute illness or surgery) (package insert). DKA developing during hospitalizations has been described emphasizing the need for vigilance when continuing SGLT-2 inhibitors in patients admitted to the hospital (192). Patients should be educated regarding this potential complication and in high-risk patients (for example patients on insulin therapy with a history of poor glycemic control or DKA) one could provide the patient with methods to measure ketone levels at home to facilitate the early diagnosis of DKA.
A possible mechanism for the increased risk of DKA is SGLT2 inhibitors increasing plasma glucagon levels thereby increasing ketone production (127,191). In combination with the low insulin levels this could potentiate the development of DKA.
OSTEOPOROSIS AND FRACTURES
In the CANVAS cardiovascular outcome study, the rate of all fractures was higher in the canagliflozin group than in the placebo group (15.4 vs. 11.9 participants with fracture per 1000 patient-years; hazard ratio, 1.26; 95% CI, 1.04 to 1.52) (133). A similar trend was observed for low-trauma fracture events (canagliflozin 11.6 vs. placebo 9.2 participants with fracture per 1000 patient-years; hazard ratio, 1.23; 95% CI, 0.99 to 1.52) (133). The incidence of fractures in the CANVAS study was increased with canagliflozin vs. placebo across subgroups based on sex, age, duration of Type 2 diabetes, baseline eGFR, and prior fracture history (193). Notably, the increase in fractures associated with canagliflozin treatment began within weeks of drug initiation indicating that the increased risk occurs rapidly (193).
In contrast, both the EMPA-REG, VERTIS, and DECLARE cardiovascular outcome studies did not demonstrate an increase in fractures with empagliflozin or dapagliflozin, respectively (132,143,148). Additionally, in the CREDENCE outcome study, canagliflozin did not increase fracture risk in patients with chronic kidney disease defined as an eGFR of 30 to <90 mL/min/1.73 m2 and albuminuria (ratio of albumin [mg] to creatinine [g], >300 to 5000) (141). Similarly, in a pooled analysis of 8 randomized canagliflozin studies with 5867 participants (CANVAS trial excluded) an increase in fractures was not observed (193). Moreover, in a meta-analysis of 27 randomized controlled trials with an average duration of 64 weeks that compared the efficacy and safety of SGLT2 inhibitors to a placebo in 20,895 participants there was no increased risk of fractures with SGLT2 inhibitor treatment (RR 1.02; 95% CI 0.81- 1.28) (194). Similarly, a meta-analysis of 10 large outcome studies also did not observe an increase in fractures (RR 1.03; 95% CI 0.95- 1.12) (181).
Several studies have examined the effect of SGLT2 inhibitors on bone mineral density. Canagliflozin was associated with a decrease in total hip bone mineral density over 104 weeks, (placebo-subtracted changes:100mg -0.9% and 300mg -1.2%), but did not result in changes in bone mineral density in the femoral neck, lumbar spine, or distal forearm (195). In a 2-year study dapagliflozin did not significantly affect bone mineral density at the lumbar spine, femoral neck, or total hip (196). In a 26-week study ertugliflozin also had no adverse effect on bone mineral density (197).
Thus, the evidence that SGLT2 inhibitors increase the risk of osteoporosis and fractures, with the possible exception of canagliflozin, is not very strong. One should recognize though, that the hypovolemia and hypotension could increase the risk of falls and thereby increase the risk of fractures in susceptible individuals.
AMPUTATIONS
In the CANVAS study described above, canagliflozin was associated with an increased risk of amputations (hazard ratio, 1.97; 95% CI, 1.41 to 2.75), which were primarily at the level of the toe or metatarsal (133). Amputation risk was strongly associated with baseline history of prior amputation and risk factors for amputation (peripheral vascular disease and neuropathy). The risk of amputation was low with 6.3 of participants per 1000 patients-years in the canagliflozin group having an amputation vs. 3.4 in the placebo group. The basis for the increase in amputations is unknown.
However, the EMPA-REG OUTCOME trial with empagliflozin, the DECLARE-TIMI 58 trial with dapagliflozin, and the VERTIS CV trial with ertuglifozin did not report an increase in amputations in the patients treated with an SGLT2 inhibitor (132,143,148,198). Moreover, in the CREDENCE trial, canagliflozin also did not cause an increase in amputations in the patients treated with the SLGT2 inhibitor (141). In a meta-analysis of 7 large cardiovascular/renal outcome trials described above (excluding CANVAS) there was no increased risk of amputations in the SGLT2 inhibitor treated group vs. placebo group (RR 1.09; CI 95% 0.94-1.26) (199). Given that only one of eight large randomized trials has demonstrated an increased risk of amputations it is unlikely that SGLT2 inhibitors significantly increase the risk of amputations.
Nevertheless, before initiating SGLT2 inhibitor therapy one should consider factors in the patient history that may predispose them to the need for amputations, such as a history of prior amputation, peripheral vascular disease, severe neuropathy, and diabetic foot ulcers and weigh the risks and benefits of therapy (package insert).
ACUTE ILLNESS
Because of the risk of hypovolemia, hypotension, and DKA the administration of SGLT2 inhibitors should be suspended during acute illness or planned surgical procedures. SGLT2 inhibitor therapy may be resumed following recovery.
This view needs to be modified based on the results of the DARE 19 study (200). In this study patients hospitalized with COVID-19 and with at least one cardiometabolic risk factor (i.e., hypertension, type 2 diabetes, atherosclerotic cardiovascular disease, heart failure, and chronic kidney disease) were randomized to dapagliflozin 10 mg daily or placebo for 30 days. While dapagliflozin did not result in a statistically significant risk reduction in organ dysfunction or death, or improvement in clinical recovery, the drug was well tolerated indicating that SGLT2 inhibitors can be safely given to hospitalized patients if there are strong indications for their use.
Contraindications and Drug Interactions
RENAL FUNCTION
The dose of SGLT2 inhibitors needs to be adjusted based on renal function. Therefore, renal function needs to be assessed prior to initiating therapy and periodically thereafter.
Dosage recommendations for dapagliflozin and canagliflozin are shown in tables 16 and 17.
|
Table 16. Dose Recommendations for Dapagliflozin
|
|
eGFR > 45
|
To improve glycemic control, the recommended starting dose is 5 mg orally once daily. Dose can be increased to 10 mg orally once daily for additional glycemic control*. For all other indications, the recommended starting dose is 10 mg orally once daily.
|
|
eGFR 25-45
|
10 mg orally once daily*
|
|
eGFR < 25
|
Initiation is not recommended; however, patients may continue 10 mg orally once daily to reduce the risk of eGFR decline, ESKD, CV death, and heart failure.
|
|
Dialysis
|
Contraindicated
|
|
Table 17. Dose Recommendations for Canagliflozin
|
|
eGFR > 60
|
100 mg orally once daily, taken before the first meal of the day. Dose can be increased to 300 mg once daily for additional glycemic control.
|
|
eGFR 30-60
|
100 mg once daily.
|
|
eGFR < 30
|
Initiation is not recommended, however patients with albuminuria greater than 300 mg/day may continue 100 mg once daily to reduce the risk of ESKD, doubling of serum creatinine, CV death, and hospitalization for heart failure
|
|
Dialysis
|
Contraindicated
|
Empagliflozin is not recommended for glycemic control in patients with an eGFR < 30 and is contraindicated in patients on dialysis. Data are insufficient to provide a dosing recommendation in patients who have type 2 diabetes and established cardiovascular disease with an eGFR less than 30 or who have heart failure with reduced ejection fraction with an eGFR less than 20.
Ertugliflozin is not recommended in patients with an eGFR less than 45 and is contraindicated in patients on dialysis.
Summary
SGLT2 inhibitors are effective at lowering glucose levels and even more importantly have beneficial effects on heart failure and renal disease. They have a number of potential side effects but many are not definitively associated with SGLT2 inhibitors (fractures, urinary tract infections, amputations) or are rare (DKA, Fournier’s gangrene). The major side effect is genital mycotic infections, which usually are mild and respond to treatment. In patients with pre-existing cardiovascular disease, at high risk for cardiovascular disease particularly heart failure, or with renal disease SGLT2 inhibitors are a leading therapeutic choice.
|
Table 18. Advantages and Disadvantages of SGLT2 Inhibitors
|
|
Advantages
|
Disadvantages
|
|
Weight loss
|
Urinary Tract Infections?
|
|
No hypoglycemia
|
Genital Mycotic Infections
|
|
Decrease heart failure
|
Increased LDL (small increase)
|
|
Decreases renal dysfunction
|
Increased risk of DKA
|
|
Once a day administration
|
Postural hypotension/volume depletion
|
|
Decrease BP
|
Fractures/ Osteoporosis?
|
|
|
Increased risk amputations (canagliflozin)?
|
|
|
Fournier’s gangrene (rare)
|
|
|
Expensive
|
COMBINATION SGLT1 AND SGLT2 INHIBITORS
Introduction
Sotagliflozin (Zynquista) inhibits both SGLT1 and SGLT2 (201). Sotaglifozin’s effectiveness in inhibiting SGLT-2 is similar to that of the selective SGLT-2 inhibitors discussed above but it is > 10-fold more potent in inhibiting SGLT-1(202). Sotagliflozin was approved in Europe for the treatment of patients with type 1 diabetes but is no longer available. In the US the drug was not approved. It was used in overweight patients (BMI> 27 kg/m2) when optimal insulin on its own does not achieve adequate glycemic control (package insert- https://www.ema.europa.eu/en/documents/product-information/zynquista-epar-product-information_en.pdf).
Administration
The recommended dose of sotagliflozin was 200mg once a day before the first meal of the day. After 3 months, the dose may be increased to 400mg once a day if additional blood sugar control is needed (package insert).
Because of an increased risk of diabetic ketoacidosis precautions should be taken to reduce this potential side effect. It is recommended that patients obtain several baseline blood or urine ketone levels over one to two weeks prior to initiation of sotagliflozin therapy and patients should become familiar with how their behaviors and circumstances affect their ketone levels. During the initial one to two weeks of treatment with sotagliflozin, ketones should be monitored on a regular basis. Measurement of blood ketone levels is preferred over urine (package insert).
In order to avoid hypoglycemia with the first dose of sotagliflozin a 20% reduction in the first mealtime bolus insulin may be considered (package insert). Subsequent bolus doses should be adjusted individually based on blood glucose results. No reduction in basal insulin is recommended when initiating sotagliflozin. Subsequently, basal insulin should be adjusted based on blood glucose results. When needed, insulin dose reduction should be done cautiously to avoid ketosis and DKA (package insert).
Mechanism of Action
The mechanism by which inhibition of SGLT2 decreases glucose levels was discussed in the prior section on SGLT2 inhibitors. Inhibition of SGLT1 will have additional effects. In the kidney SGLT1 is responsible for approximately 10% of the transport of luminal glucose and thus inhibiting SGLT1 may facilitate SGLT2 induced loss of glucose in the urine (201,203). Moreover, SGLT1 is expressed in the small intestine and facilitates the absorption of dietary glucose (201,203,204). SGLT1 expression in the small intestine is increased in patients with diabetes (203,204). Inhibition of SGLT1 delays, and perhaps reduces, glucose absorption, and enhances circulating levels of GLP-1 reducing post-prandial glucose excursions (201,204-206). Finally, SGLT1 is expressed in human heart capillaries and whether this plays a role in cardiac protection remains to be determined (202).
Glycemic Efficacy
TYPE 1 DIABETES (T1DM)
The inTandem1 trial was carried out in North American adults and randomized patients with T1DM to placebo (n = 268), sotagliflozin 200 mg (n = 263), or sotagliflozin 400 mg (n = 262) (207). Baseline A1c was 7.57% and the placebo-adjusted A1c reductions were 0.36% and 0.41% with sotagliflozin 200 and 400 mg, respectively, at 24 weeks and 0.25% and 0.31% at 52 weeks (all P < 0.001). At 52 weeks the difference in body weight between the placebo group and 400mg sotagliflozin group was -4.32 kg (-5.00 to -3.64). Notably hypoglycemia was not increased with sotagliflozin treatment. However, DKA occurred more frequently with sotagliflozin treatment (placebo 0.4%, sotagliflozin 200mg 3.4%, sotagliflozin 400mg 4.2%).
The inTandem2 trial was carried out in European adults and randomized patients with T1DM to placebo (n = 258), oral sotagliflozin 200 mg (n = 261), or 400 mg (n = 263) (208). Baseline A1c was 7.7% and the placebo-adjusted A1c reductions were 0.37% and 0.35% with sotagliflozin 200 and 400 mg, respectively, at 24 weeks and 0.21% and 0.37% at 52 weeks (all P < 0.001). At 52 weeks the difference in body weight between the placebo group and 400mg sotagliflozin group was −2.92 kg (-3.62 to −2.22). Hypoglycemia was not increased with sotagliflozin treatment. DKA occurred more frequently with sotagliflozin treatment (placebo 0%, sotagliflozin 200mg 2.3%, sotagliflozin 400mg 3.4%).
The inTandem3 trial was a multicenter world-wide study in patients with T1DM randomized to placebo (n=703) or sotagliflozin 400mg (n=699) for 24 weeks (209). The baseline A1c was 8.2% and sotagliflozin decreased A1 by −0.46% compared to placebo. Hypoglycemia with a blood glucose level < 55 mg/dL was significantly lower in the sotagliflozin group than in the placebo group (11.8 per person-year vs. 15.4 per person-year) but severe hypoglycemia (episode needing assistance from another person or resulting in loss of consciousness or a seizure) was similar. Notably the risk of DKA was increase with sotagliflozin treatment (sotagliflozin 3.0% and placebo 0.6%).
Thus, in patients with T1DM sotagliflozin causes a modest reduction in A1c and body weight but increases the risk of DKA.
TYPE 2 DIABETES
Studies of the effect of sotagliflozin on glycemic control in patients with T2DM have not been as extensive as in patients with T1DM. In a 12-week trial that compared placebo (n= 60), canagliflozin 200mg (n= 60), or sotagliflozin 400mg (n= 60) in patients with T2DM on metformin monotherapy a decrease in A1c of -0.09%, -0.50, and -0.92% occurred in patients treated with placebo, sotagliflozin 200mg, and sotagliflozin 400mg, respectively (210). As expected, there was a decrease in body weight and an increase in urinary glucose excretion with sotagliflozin treatment. Of note a study has shown that in patients with T2DM sotagliflozin treatment is effective in lowering postprandial glucose levels even in patients with an eGFR < 45 mL/min/1.73 m2 (211).
Other Effects
CARDIOVASCULAR
The SOLOIST-WHF Trial was a multicenter trial in which patients with T2DM who were recently hospitalized for worsening heart failure were randomly assigned to receive sotagliflozin 200 mg once daily (with a dose increase to 400 mg, depending on side effects) (n= 608), or placebo (n= 614) (212). The primary end point was the total number of deaths from cardiovascular causes and hospitalizations and urgent visits for heart failure (first and subsequent events). Because of loss of funding from the sponsor the study was stopped early and the median duration of follow-up was 9 months. The primary end-point was reduced in the sotagliflozin group vs. placebo group (HR 0.67; 95% CI, 0.52 to 0.85; P<0.001) as was hospitalizations or urgent visits for heart failure (HR 0.64; 95% CI, 0.49 to 0.83: P <0.001). Of particular note benefit was observed in patients with reduced or preserved ejection fractions (<50% or ≥50%). This study demonstrates benefits in patients with a reduced or preserved ejection fractions and that treatment initiated during an acute heart failure episode is beneficial. DKA was uncommon in both the sotagliflozin group (0.3%) and placebo group (0.7%) but severe hypoglycemia was increased (sotagliflozin 1.5% vs placebo 0.3%).
The SCORED trial was a multicenter trial in which patients with T2DM and chronic kidney disease (eGFR- 25 to 60 ml/min/1.73 m2, albuminuria was not required), and risks for cardiovascular disease were randomized to sotagliflozin (200 mg once daily, with an increase to 400 mg once daily if unacceptable side effects did not occur) (n= 5292) or placebo (n= 5292) and followed for a median of 16 months (213). The primary end point was the composite of the total number of deaths from cardiovascular causes, hospitalizations for heart failure, and urgent visits for heart failure. Sotagliflozin treatment decreased the primary end point (HR 0.74; 95% CI, 0.63–0.88; P <0.001), hospitalizations or urgent visits for heart failure (HR 0.67; 95% CI, 0.55–0.82; P <0.001), and deaths from cardiovascular causes, nonfatal myocardial infarctions, and nonfatal strokes (HR 0.77; 95%CI 0.65–0.91). There was a trend towards a reduction in a renal composite endpoint consisting of a decrease of ≥50% in the eGFR from baseline for ≥30 days, long-term dialysis, renal transplantation, or sustained eGFR of <15 ml/min/1.73 m2 for ≥30 days (HR 0.71; 95% CI, 0.46–1.08). DKA while infrequent was increased in the sotagliflozin group (0.6% vs 0.3%; P=0.02).
RENAL
While outcome studies are not available in patients with T1DM it has been shown that sotagliflozin has effects on kidney function that are similar to what has been observed in patients with T2DM. Specifically, there was an acute decrease in eGFR and a decrease in albuminuria (214). These observations suggest that the beneficial effects on renal outcomes seen with SGLT2 inhibitors in patients with T2DM and non-diabetics will also occur in patients with T1DM.
Side Effects
The side effects of sotagliflozin are similar to those described previously for SGLT2 inhibitors. In addition, sotagliflozin also causes diarrhea and flatulence due to the inhibition of SGLT1 mediated glucose uptake in the small intestine.
Contraindications and Drug Interactions
Patients at high risk for DKA should not be started on sotagliflozin.
Summary
In patients with T1DM sotagliflozin modestly reduces A1c levels and body weight but increases the risk of DKA. Because of the increased risk of DKA this drug is no longer approved for use.
In patients with T2DM sotagliflozin use is not approved in the US or Europe. While studies have shown beneficial effects of sotagliflozin on cardiovascular disease it is not clear whether this benefit is solely due to inhibition of SGLT2 or whether inhibition of SGLT1 plays a significant role.
DOPAMINE AGONIST (CYCLOSET)
Introduction
In 2009, a quick-release formulation of bromocriptine (Cycloset, bromocriptine-QR) was approved to improve glycemic control in patients with T2DM (215,216). Bromocriptine is a centrally-acting dopamine D2 receptor agonist that has been used for many years for the treatment of hyperprolactinemia and Parkinson’s disease (215,216). It can be used to improve glycemic control in patients with T2DM either as monotherapy or in combination with other hypoglycemic drugs (215,216)
Administration
Bromocriptine-QR should be initiated at one tablet (0.8 mg) within two hours after waking in the morning. The dose can be increased by one tablet per week until a maximum daily dose of 6 tablets (4.8 mg) or until the maximal tolerated number of tablets between 2 and 6 per day is reached. Taking bromocriptine-QR with food is recommended to decrease gastrointestinal side effects (215).
Mechanism of Action
Bromocriptine-QR decreases insulin resistance resulting in an increase in glucose disposal and a decrease in hepatic glucose production (215). Bromocriptine-QR does not increase insulin levels (215). Thus, the effectiveness of bromocriptine-QR will be greatest in patients that are insulin resistant and produce insulin (215). Based on animal studies it is thought that bromocriptine-QR acts on the central nervous system, particularly the hypothalamus, to increase insulin sensitivity in liver, muscle, and adipose tissue (215).
Glycemic Efficacy
In a 24 week monotherapy study the A1c level was 0.4% lower in the bromocriptine-QR group compared to placebo group (217). Both fasting and postprandial glucose levels were decreased with bromocriptine-QR treatment (217). Bromocriptine-QR treatment was associated with a decrease in triglyceride levels (32 mg/dL) but no significant change in LDL or HDL cholesterol levels or change in body weight (217). A trial adding bromocriptine-QR to sulfonylurea therapy demonstrated a 0.55% lower A1c in the bromocriptine-QR group compared to placebo (217). As in the monotherapy study fasting glucose, postprandial glucose, and triglyceride levels were decreased with no change in LDL or HDL cholesterol levels (217). Addition of bromocriptine-QR to other hypoglycemic drugs including insulin results in an approximate decrease in A1c of 0.5 to 1.0% (215,216). Hypoglycemia is a rare side effect with use of bromocriptine-QR alone, but is increased with use of insulin secretagogue therapy or insulin (216,217).
Other Effects
BLOOD PRESSURE
Bromocriptine-QR modestly decreases systolic and diastolic blood pressure (216,217).
LIPIDS
Bromocriptine-QR treatment decreases triglyceride levels but has no significant effect on LDL or HDL cholesterol levels (216,217). The decrease in triglyceride levels is thought to be due to a decrease in hepatic triglyceride synthesis, likely due to a decrease in adipose tissue lipolysis resulting in decreased blood free fatty acid levels and decreased delivery of fatty acids to the liver for triglyceride synthesis (215).
CARDIOVASCULAR DISEASE
A 52-week, randomized, double-blind, multicenter trial evaluated cardiovascular safety in 3,095 patients with T2DM treated with bromocriptine-QR or placebo (218). The composite end point of first myocardial infarction, stroke, coronary revascularization, or hospitalization for angina or congestive heart failure occurred in 1.8% of the bromocriptine-QR treated vs. 3.2% of the placebo-treated patients resulting in a 40% decrease in cardiovascular events (HR 0.60; CI 0.37– 0.96). Clearly further studies to confirm this finding and to elucidate the mechanism of this beneficial effect are required.
Side Effects
The most common side effect of bromocriptine-QR therapy is nausea which is usually transient and improves with time (216,217). This side effect can be minimized by reducing the dose (216,217). In the pooled phase 3 trials adverse events leading to discontinuation occurred in 539 (24%) of the bromocriptine-QR treated patients and 118 (9%) of the placebo-treated patients. This between-group difference was driven mostly by gastrointestinal adverse events, particularly nausea (package insert). Similarly, in the bromocriptine-QR safety trial adverse events leading to discontinuation of drug occurred in 24% of the bromocriptine-QR treated patients and 15% of the placebo-treated patients, a difference again driven mostly by gastrointestinal adverse events, particularly nausea (package insert).
Hypotension resulting in syncope can occur particularly in patients on anti-hypertensive medications (package insert). Other side effects include somnolence, fatigue, vomiting, headache, and dizziness (package insert).
Contraindications and Drug Interactions
Bromocriptine-QR is metabolized by the Cyp3A4 system and therefore the drug should not be used with strong CYP3A4 inhibitors (e.g., azole antimycotics, HIV protease inhibitors) and the dose should not exceed 1.6 mg once daily during concomitant use of a moderate CYP3A4 inhibitor (e.g., erythromycin) (package insert).
Bromocriptine-QR is contraindicated in patients with syncopal migraine because it increases the likelihood of a hypotensive episode (package insert). The use of bromocriptine-QR in patients with severe psychotic disorders in not recommended as it may exacerbate the disorder or diminish the effectiveness of drugs used to treat the disorder (for example clozapine, olanzapine, ziprasidone) (package insert).
Summary
Bromocriptine-QR has modest effects on A1c levels by decreasing insulin resistance. In clinical trials the drug was often discontinued due to nausea. Because of the modest effects on A1c and the prominent side effects this drug is not widely used in the treatment of patients with T2DM. If further studies confirmed the decrease in cardiovascular events in patients treated with bromocriptine-QR the use of this drug would increase.
|
Table 19. Advantages and Disadvantages of Bromocriptine-QR
|
|
Advantages
|
Disadvantage
|
|
Decreases triglycerides
|
Need to titrate dose
|
|
Once a day dosing
|
Modest effect on A1c
|
|
Cardiovascular benefits?
|
Frequent discontinuation due to GI side effects
|
|
Decrease BP
|
Expensive
|
|
Neutral weight effect
|
|
|
Hypoglycemia uncommon
|
|
OVERVIEW OF THE INCRETIN SYSTEM
The incretin effect refers to a greater insulin stimulatory effect after an oral glucose load than from an intravenous glucose infusion when plasma glucose concentrations are matched (219). Thus, glucose and other nutrients delivered via the gastrointestinal tract potentiates the ability of the beta cells in the pancreas to produce insulin resulting in greater insulin secretion than with IV glucose (220). The increase in insulin levels with IV glucose is only approximately one‐third of that elicited by oral glucose. The majority of the incretin effect is due to two GI hormones, glucose-dependent insulinotropic peptide (GIP) and glucagon like peptide-1 (GLP-1) with GIP having a dominant role (Figure 9) (219). The basal plasma levels of the incretin hormones are low but after eating the levels increase reaching concentrations that augment the insulin secretory responses if glucose levels are high but are ineffective at low glucose concentrations (i.e. glucose dependent effect) (219).
Patients with T2DM have a significant reduction of the incretin effect but GLP-1 and GIP levels in the blood after meals are not reduced in patients with T2DM (219). Rather decreased functional beta cell mass and resistance to the effects of GLP-1 and GIP in patients with T2DM accounts for the decreased incretin effect (219). Infusion of GIP has a minimal response on insulin secretion in patients with T2DM (resistance to effect of GIP) whereas GLP-1 administration is able to stimulate insulin secretion but the response is reduced in patients with T2DM compared to normal individuals likely secondary to decreased functional beta cell mass (219). Achieving near-normoglycemia by intensified insulin regimens improved beta cell responsiveness to exogenous GIP and GLP-1, although the increase in insulin secretion was still much lower than in individuals without diabetes (219). The reduced incretin effect in patients with T2DM occurs after the diagnosis of diabetes is established, suggesting this abnormality is secondary to the diabetic state rather than the cause of diabetes (220).
Glucagon Like Peptide-1 (GLP-1)
GLP-1 is cleaved from the pro-glucagon molecule by pro-hormone convertase enzymes in the intestine (220). GLP-1 is stored in the L-cells of the intestine, predominantly in the ileum and colon, and is released at mealtime in response to neurohormonal signals and the presence of food in the gut (219,220). GLP-1 affects postprandial glucose levels through several mechanisms, including enhancing insulin secretion by the beta cells and inhibiting postprandial glucagon secretion by the alpha cells in a glucose-dependent manner (i.e. GLP-1 does not stimulate insulin secretion or inhibit glucagon secretion unless glucose levels are elevated) (220). This glucose dependent effect accounts for why incretin-based drugs do not cause serious hypoglycemia. Activation of GLP-1 receptors on beta cells increases cAMP levels, which potentiates insulin release in the presence of elevated glucose concentrations. In addition, GLP-1 slows the rate of gastric emptying, which is often paradoxically accelerated in patients with diabetes (220). GLP-1 also acts as a postprandial satiety signal through neurohormonal networks that signal the brain to suppress appetite and food intake, which can lead to weight loss (220). Animal studies suggest that exogenous GLP-1 has the ability to increase islet size, enhance beta-cell proliferation, inhibit beta-cell apoptosis, and regulate islet growth (221). The administration of GLP-1 intravenously increases insulin secretion, reduces glucagon secretion, and decreases glucose levels during fasting and in the post-prandial state (219). GLP-1 is rapidly degraded by dipeptidyl peptidase 4 (DPP-4) into inactive peptides (half-life is minutes) (Figure 9).
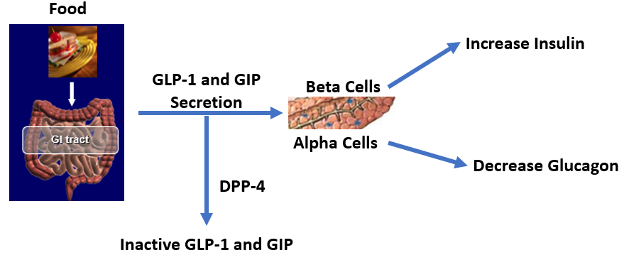
Figure 9. Incretin Hormone Secretion and Effect on Pancreas
Glucose-Dependent Insulinotropic Peptide (GIP)
Within minutes after ingestion of food, GIP is secreted from the K-cells located in the proximal region of the jejunum (219,220). GIP helps maintain normal glucose homeostasis by stimulating an increase in insulin secretion by the beta cells (Figure 9). Studies have suggested that the increase in insulin with food intake (Incretin effect) is primarily mediated by GIP (219). In contrast to GLP-1, GIP does not inhibit glucagon secretion, and in fact may stimulate glucagon secretion during euglycemic states. Additionally, GIP has no effect on gastric emptying or on satiety. GIP concentrations in patients with T2DM are either normal or slightly increased following a meal indicating that the failure to secrete is not the explanation for the decreased incretin effect. Rather, beta cells in patients with T2DM are resistant to GIP. GIP is rapidly degraded by DPP-4 into inactive peptides (half-life is minutes) (Figure 9). The characteristics of GLP-1 and GIP are shown in table 20.
|
Table 20. Characteristics of GLP-1 and GIP
|
|
|
GLP-1
|
GIP
|
|
Post meal levels in patients with diabetes
|
Normal
|
Normal
|
|
Effect on insulin secretion
|
Stimulates
|
Stimulates
|
|
Effect on glucagon secretion
|
Inhibits
|
No effect or stimulates
|
|
Gastric emptying
|
Delays
|
No effect
|
|
Satiety
|
Induces
|
No effect
|
|
Degradation by DPP-4
|
Yes
|
Yes
|
DIPEPTIDYL PEPTIDASE-4 (DPP-4) INHIBITORS
Introduction
The currently available DPP-4 inhibitors in the US are sitagliptin (Januvia), saxagliptin (Onglyza), linagliptin (Tradjenta), and alogliptin (Nesina). Vidigliptin (Galvus) is available in Europe (222). DPP-4 inhibitors can be used as monotherapy, dual therapy, triple drug therapy, or in combination with insulin (222). These drugs are very similar and the minor differences will be discussed below.
Administration
The recommended dose of sitagliptin is 100 mg once daily with or without food. In patients with moderate renal impairment (eGFR greater than or equal to 30 mL/min/1.73 m2 but less than 45 mL/min/1.73 m2), the dose of sitagliptin is 50 mg once daily. In patients with severe renal impairment (eGFR less than 30 mL/min/1.73 m2) the dose of sitagliptin is 25 mg once daily.
The recommended dosage of saxagliptin is 2.5 mg or 5 mg once daily with or without food. In patients with a creatinine clearance CrCl ≤50 mL/min the dose of saxagliptin is 2.5 mg.
The recommended dose of linagliptin is 5 mg once daily with or without food. No dose adjustment is required for decreased renal function.
The recommended dose of alogliptin is 25 mg once daily with or without food. The dose of alogliptin is 12.5 mg once daily for patients with moderate renal impairment (CrCl ≥30 to <60 mL/min) and 6.25 mg with severe renal impairment (CrCl <30 mL/min).
Renal function should be checked prior to initiating treatment and periodically because dose adjustments are required for all DPP-4 inhibitors except linagliptin.
Mechanism of Action
DPP-4 inhibitors increase the concentration and activity of the endogenous incretins, GLP-1 and GIP, by inhibiting the proteolytic cleavage of these hormones by DPP-4, into inactive molecules (222). As discussed above, GLP-1 is secreted by L-cells in the intestines and stimulates insulin secretion and suppresses glucagon secretion in a glucose dependent manner, inhibits gastric emptying, and has central anorexic activity that decreases food intake. GIP is secreted by the K cells in the proximal intestine and stimulates insulin secretion in a glucose dependent manner.
An increase in active GLP-1 and GIP potentiates glucose induced insulin secretion and an increase in GLP-1 inhibits glucagon secretion (222). Together an increase in insulin and a decrease in glucagon will result in a decrease in blood glucose levels. Of note, DPP-4 inhibition results in a 2–3-fold increase in postprandial active GLP-1 levels, which is not at a level that delays gastric emptying or increases satiety and induces weight loss. This is in contrast to GLP-1 receptor agonist administration that results in marked elevations in active GLP1 activity that is equivalent to a >10-fold increase in GLP-1, which can delay gastric emptying and increase satiety.
Glycemic Efficacy
DPP-4 inhibitors typically reduce A1c levels by 0.5-1.0% and are less effective in lowering A1c compared to metformin, TZDs, SGLT2 inhibitors, and GLP-1 receptor agonists (Table 6) (12,16,222). With regards to sulfonylureas, studies have shown a greater decrease in A1c with sulfonylureas compared to DPP-4 inhibitors in short term studies but in studies greater than one year the effect of sulfonylureas and DPP-4 inhibitors on A1c were similar (16,222). The ability of DPP-4 inhibitors to lower A1c is similar in monotherapy and when DPP-4 inhibitors are used in combination with other drugs (16,222). The decrease in A1c is similar for the different DPP-4 inhibitors (12,16). DPP-4 inhibitors are effective in lowering postprandial glucose levels. Because of their mechanism of action, DPP-4 inhibitors do not cause hypoglycemia but can potentiate the hypoglycemia induced by insulin or sulfonylureas (16,222). An adjustment in the dose of sulfonylureas or insulin may be required to reduce the risk of hypoglycemia.
Other Effects
WEIGHT
DPP-4 inhibitors are weight neutral (16,222).
BLOOD PRESSURE
A meta-analysis of 15 trials involving 5,636 participants found that DPP-4 inhibitors compared to placebo reduced systolic BP (mean difference, -3.04 mmHg: P < 0.00001) and diastolic BP (mean difference, -1.47 mmHg; P < 0.00001) (223).
LIPIDS
DPP-4 inhibitors decrease postprandial triglycerides by reducing circulating chylomicrons by decreasing intestinal lipoprotein production while having minimal effects on fasting lipid levels (224).
CARDIOVASCULAR DISEASE
The effect of the DPP-4 inhibitors saxagliptin, alogliptin, sitagliptin, and linagliptin on cardiovascular endpoints has been reported. In the saxagliptin study (SAVOR‐TIMI 53 trial), 16,492 patients with T2DM who had a history of cardiovascular events or who were at high risk were randomized to saxagliptin or placebo for 2.1 years (225). Saxagliptin did not increase or decrease cardiovascular death, myocardial infarction, or ischemic stroke. Interestingly more patients treated with saxagliptin were admitted to the hospital for heart failure. The risk of heart failure with saxagliptin was greatest in patients at a high overall risk of heart failure (i.e., history of heart failure, impaired renal function, or elevated baseline levels of NT-proBNP) (226). Additionally, in the patients treated with saxagliptin the increase in heart failure was an early event with a 6-month rate of 1.1% vs. 0.6% in the placebo group (HR 1.80, p=0·001) and a 12-month rate of 1·9% vs. 1·3% (1.46; p=0.002) (226). In contrast, after 12 months no difference in the rate of heart failure was observed in the saxagliptin and placebo groups indicating that the development of heart failure is an early event (226)
In the alogliptin trial (EXAMINE), 5,380 patients with either an acute myocardial infarction or unstable angina within the previous 15-90 days were randomized to alogliptin or placebo and followed for a median of 18 months (227). As seen in the saxagliptin study the rates of cardiovascular events (death from cardiovascular causes, non-fatal myocardial infarction, or non-fatal stroke) were similar in the alogliptin and placebo groups. The risk of hospitalization for heart failure was not statistically increased in the entire subset of patients treated with alogliptin (228). However, the hazard ratio for the subgroup of patients without heart failure at baseline was 1.76, p=0.026) (228).
In the sitagliptin trial (TECOS), 14,671 patients with established cardiovascular disease were randomized to sitagliptin or placebo for 3 years (229). Sitagliptin did not decrease the risk of major adverse cardiovascular events or increase hospitalization for heart failure. Finally, in the linagliptin trial (CARMELINA), 6,979 patients at high risk for cardiovascular disease were randomized to linagliptin or placebo for a median follow-up of 2.2 years (230). As in the other DPP-4 inhibitor studies, linagliptin did not have a beneficial effect on cardiovascular events. Additionally, linagliptin did not increase the risk of hospitalization for heart failure (231).
Thus, these results indicate that DPP-4 inhibitors do not reduce cardiovascular disease. Whether specific DPP-4 inhibitors (saxagliptin, alogliptin) increase the risk of heart failure remains to be resolved. Of note, a meta-analysis of 30 randomized controlled trials involving 29,938 patients comparing the effects of saxagliptin vs. placebo or sulfonylureas did not observe an increase in heart failure (RR 0.99, 95% CI 0.89 to 1.10; p = 0.85) (232).
RENAL DISEASE
Changes in renal function were examined in the large cardiovascular outcome trials described above. In the SAVOR-TIMI 53 trial treatment with saxagliptin decreased albuminuria but had no effect on eGFR (233). Saxagliptin reduced the development of macroalbuminuria independent of changes in A1c levels (225,233). Doubling of serum creatinine, initiation of chronic dialysis, renal transplantation, or serum creatinine >6.0 mg/dL, were similar in the saxagliptin and placebo groups (233). In the TECOS trial treatment with sitagliptin also reduced the urinary albumin to creatinine ratio with no effect on eGFR (234). In the CARMELINA trial many of the patents had pre-existing renal disease (74% of patients had prevalent diabetic kidney disease, 43% had an eGFR below 45 mL/min/1.73 m2, 15.2% had an eGFR below 30 mL/min/1.73 m2 and 80% had a urinary albumin creatinine ratio >30 mg/g) (230). Treatment with linagliptin reduced the progression of albuminuria but had no effect on death due to renal failure, ESRD, or sustained 40% or higher decrease in eGFR from baseline (230).
Taken together these studies indicate that DPP-4 inhibitors decrease proteinuria but do not provide data suggesting an improvement or delay in worsening of renal function.
Side Effects
DPP-4 inhibitors have been safe drugs with minimal side effects and are well tolerated by patients. Very rarely hypersensitivity reactions including urticaria, facial edema, anaphylaxis, angioedema, and exfoliative skin conditions including Stevens-Johnson syndrome have occurred (package insert). Bullous pemphigoid has also rarely been associated with DPP-4 inhibitor treatment (package insert).
ACUTE PANCREATITIS
The package insert of DPP-4 inhibitors indicates that acute pancreatitis is a complication of DPP-4 inhibitor treatment. The individual results of the SAVOR–TIMI, EXAMINE, and TECOS trials discussed above did not show an increased risk of pancreatitis or pancreatic cancer. However, two meta-analyses of these studies demonstrated an 80% increased risk of acute pancreatitis in patients using DPP-4 inhibitors compared with those receiving standard care (235,236). It should be noted that the absolute risk was small (0.13%), which would result in one to two additional cases of acute pancreatitis for every 1,000 patients treated for 2 years (236). Thus, pancreatitis appears to be a rare side effect of DPP-4 inhibitors. In patients on DPP-4 inhibitors who have GI symptoms suggestive of pancreatitis further evaluation is indicated. The diagnosis of acute pancreatitis requires the presence of two of the following three criteria: acute onset of persistent, severe, epigastric pain often radiating to the back, elevation in serum lipase or amylase to three times or greater than the upper limit of normal, and characteristic findings of acute pancreatitis on imaging (237).
ARTHRALGIA
Severe and disabling arthralgia in patients taking DPP-4 inhibitors has been reported (238). The time to onset of symptoms following initiation of drug therapy varied from one day to years. Patients experienced relief of symptoms upon discontinuation of the medication and a subset of patients experienced a recurrence of symptoms when restarting the same drug or a different DPP-4 inhibitor. If a patient develops severe joint pain discontinue the DPP-4 inhibitor.
Contraindications and Drug Interactions
It is unknown whether patients with a history of pancreatitis or who are at increased risk for the development of pancreatitis should be started on DPP-4 inhibitors. Given the availability of other hypoglycemic drugs many clinicians avoid the use of DPP-4 inhibitors in these patients.
The dosage of saxagliptin is 2.5 mg once daily when co-administered with a strong cytochrome P450 3A4/5 inhibitor (e.g., ketoconazole, atazanavir, clarithromycin, indinavir, itraconazole, nefazodone, nelfinavir, ritonavir, saquinavir, and telithromycin) (package insert).
Summary
DPP-4 inhibitors, while not the most potent drugs at lowering A1c, nevertheless are very attractive to use in the treatment of patients with T2DM as they are safe drugs that do not have many side effects. They do not cause hypoglycemia, weight gain, or cardiovascular disease. Unfortunately they do not reduce the risk of cardiovascular disease or prevent loss of renal function.
|
Table 21. Advantages and Disadvantages of DPP-4 Inhibitors
|
|
Advantages
|
Disadvantages
|
|
No hypoglycemia
|
Pancreatic disease
|
|
Weight neutral
|
Heart failure (saxagliptin/alogliptin)?
|
|
Decreases postprandial glucose
|
Arthritis
|
|
Once a day
|
Bullous pemphigoid
|
|
Well tolerated
|
Relatively expensive
|
|
Decreases BP
|
Modest glycemic lowering
|
INJECTABLE GLUCAGON LIKE PROTEIN-1 (GLP-1) RECEPTOR AGONISTS
Introduction
There are currently six GLP-1 receptor agonists available in the US, three drugs administered daily and three drugs administered weekly (Figure 10). Albiglutide (Tanzeum) was withdrawn from the market for commercial reasons and is no longer available. GLP-1 receptor agonists can be used in combination with multiple oral anti‐diabetic drugs or in combination with insulin (239). The concentrations of GLP-1 receptor agonist activity are much higher than physiological levels of GLP-1 activity (16). The GLP-1 receptor agonists that a similar to exendin-4 (Exenatide and Lixisenatide) are eliminated by the kidneys and therefore in patients with severe renal disease these drugs are contraindicated (16). In contrast, the drugs that are analogues of GLP-1 are degraded by peptidases (16).
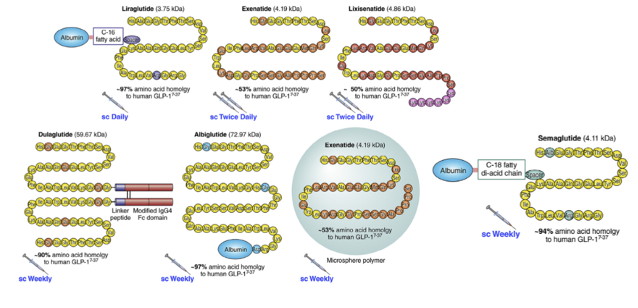
Figure 10. Structure of GLP-1 Receptor Agonists
SHORT ACTING GLP-1 RECEPTOR AGONISTS
Exenatide (Byetta) is a synthetic exendin-4 that is a peptide originally isolated from the saliva of the Gila monster that has a 53% homology with human GLP-1 and is resistant to degradation by DPP-4 (16,239). Lixisenatide (Adylyxin) is an exendin-4 analogue with six Lys residues added at the C terminus to confer resistance to DPP-4 (16,239).
LONG ACTING GLP-1 RECPTOR AGONISTS
Even though liraglutide (Victoza) is administered daily it is considered a long acting GLP-1 receptor agonist because its effects on fasting glucose levels are similar to weekly GLP-1 receptor agonists and its effects on gastric emptying wane as seen with weekly GLP-1 receptor agonists. Liraglutide is an analogue of GLP-1 with the addition of a 16-carbon fatty acid chain that masks the DPP-4 cleavage site preventing degradation (8,179). Once weekly exenatide (Bydureon and Bydueron BCise) is a sustained-release formulation that consists of exenatide embedded within biodegradable polymeric microspheres of poly (DL-lactic-co-glycolic acid) (16). Dulaglutide (Trulicity) has two copies of a GLP-1 analogue covalently linked to an Fc fragment of human IgG4 (16,239). Semaglutide (Ozempic) is an analogue of human GLP‐1 RA and is linked via a hydrophilic spacer and a fatty acid side chain to albumin (239).
Administration
SHORT ACTING GLP-1 RECEPTOR AGONISTS
Initiate exenatide at 5 ug twice daily; increase to 10 ug twice daily after 1 month based on clinical response. Inject subcutaneously within 60 minutes prior to morning and evening meals (or before the two main meals of the day).
The starting dose of lixisenatide is 10 ug subcutaneously once daily within one hour before the first meal of the day for 14 days and then increase the dose to the maintenance dose of 20 ug once daily.
LONG ACTING GLP-1 RECPTOR AGONISTS
Initiate liraglutide with a dose of 0.6 mg per day for one week. After one week at 0.6 mg per day, the dose should be increased to 1.2 mg. If the 1.2 mg dose does not result in acceptable glycemic control, the dose can be increased to 1.8 mg. Inject subcutaneously once-daily at any time of day, independently of meals.
The recommended dose of long acting exenatide is 2 mg subcutaneously once every 7 days (weekly). The dose can be administered at any time of day, with or without meals.
The recommended initiating dose of dulaglutide is 0.75 mg subcutaneously with or without food once weekly. The dose may be increased to 1.5 mg once weekly to achieve glycemic control. If after 4 weeks glycemic control is not achieved the dose can be increased to 3.0mg once weekly and then after another 4 weeks to 4.5mg once weekly for additional glycemic control.
The recommended initiating dose of semaglutide is 0.25 mg subcutaneous injection with or without food once weekly for 4 weeks. The 0.25 mg dose is intended for treatment initiation and is not effective for glycemic control. After 4 weeks on the 0.25 mg dose, increase the dosage to 0.5 mg once weekly. If additional glycemic control is needed after at least 4 weeks on the 0.5 mg dose, the dosage may be increased to 2 mg once weekly.
Note that exenatide and lixisenatide are contraindicated in patients with renal dysfunction (for details see Contraindications section).
Information on the pen delivery systems for the GLP-1 receptor agonists is shown in table 22.
|
Table 22. Characteristics of GLP-1 Receptor Agonist Pen Devices
|
|
Generic
|
Exenatide
|
Exenatide
|
Exenatide
|
Lixisenatide
|
Liraglutide
|
Dulaglutide
|
Semaglutide
|
|
Brand
|
Byetta
|
Bydureon
|
Bydureon
BCise
|
Lyxumia
|
Victoza
|
Trulicity
|
Ozempic
|
|
Single or multiple use
|
Multiple
|
Single
|
Single
|
Multiple
|
Multiple
|
Single
|
Multiple
|
|
Dose*
|
5 or 10ug
|
2mg
|
2mg
|
10 or 20ug
|
0.6, 1.2, or 1.8mg
|
0.75 or 1.5mg
|
0.25, 0.5, or 1.0mg
|
|
Preparation
|
None
|
Resuspend
|
Mix
|
None
|
None
|
None
|
None
|
*Only the liraglutide pen can deliver different doses
Mechanism of Action
GLP-1 receptor agonists potentiate glucose dependent insulin secretion increasing insulin levels and lowering glucose levels (16). In addition, GLP-1 receptor agonists potentiate the glucose dependent inhibition of glucagon secretion, which will also lower glucose levels (16). Finally, because of the supraphysiological levels of GLP-1 activity, short-acting GLP-1 receptor agonists will delay gastric emptying resulting in a decrease in postprandial glucose levels and induce satiety, which will decrease food intake (16).
Glycemic Efficacy
GLP-1 receptor agonists typically lower A1c by 1-2% (16). The efficacy of GLP-1 receptor agonists vary with semaglutide being the most potent and lixisenatide being the least potent (see table 6) (12). Note table 6 does not include the 3.0mg and 4.5mg of dulaglutide, which lower A1c by 1.6% and 1.8% respectively (240). In general, long acting GLP-1 receptor agonists are better at lowering A1c levels compared to short acting agents (12,239). The efficacy in lowering A1c is similar in monotherapy and during combination therapy (16). The reduction in A1c is sustained over several years (164). Long acting GLP-1 receptor agonists lower fasting glucose levels more effectively than short acting drugs (239). Conversely, short acting GLP-1 receptor agonists lower postprandial glucose excursions to a greater extent than long acting agents (239). Short acting GLP-1 receptor agonists induce a substantial retardation in gastric emptying, which likely contributes significantly to the lowering of postprandial glucose excursions after meals when they are administered (239). Notably, the ability of short acting GLP-1 receptor agonists to prevent postprandial glucose excursions is greatly diminished for meals when they are not administered (239). In patients with diminished beta cell function the glycemic response to GLP-1 receptor agonist therapy is reduced (241).
Studies have compared adding a GLP-1 receptor agonist to basal insulin vs. adding rapid acting insulin to basal insulin (242). In a meta-analysis there were no differences in lowering A1c levels but treatment with basal insulin plus GLP-1 receptor agonist led to a significant reduction in body weight, whereas basal insulin plus rapid acting insulin treatment was associated with weight gain (difference -2.95 kg; p = 0.0001) (242). Additionally, patients treated with basal insulin plus GLP-1 receptor agonist were less likely to experience symptomatic hypoglycemia (OR: 0.52; p < 0.0001) and severe hypoglycemia (OR: 0.27; p = 0.07) than those treated with basal insulin plus rapid acting insulin. Thus, adding a GLP-1 receptor agonist to basal insulin instead of bolus insulin will result in similar improvements in glycemic control with fewer side effects.
Studies have also compared adding insulin therapy vs. adding a GLP-1 receptor agonist. In a meta-analysis of 19 studies GLP-1 receptor agonists reduced A1c levels slightly more than insulin therapy (difference -0.12%, P < .0001) (243). As expected, hypoglycemia was less frequent in the patients treated with the GLP-1 receptor agonists.
Because the effect of GLP‐1 receptor agonists on insulin and glucagon secretion are glucose dependent they have a low potential to cause hypoglycemia (16,239). The risk of hypoglycemia increases when these GLP-1 receptor agonists are used in combination with insulin or secretagogues (239).
Both GLP-1 receptor agonists and SGLT-2 inhibitors have been shown to decrease cardiovascular disease (GLP-1 receptor agonists decrease primarily atherosclerotic complications while SGLT-2 inhibitors primarily decrease heart failure). Therefore, the use of these drugs in combination to prevent cardiovascular disease has been proposed. In an analysis of four randomized trials adding a GLP-1 receptor agonist to a SGLT-2 inhibitor it was reported that the addition of a GLP-1 receptor agonist resulted in a greater reduction in HbA1c (-0.74%), body weight (-1.61 kg), and systolic blood pressure (-3.32 mmHg) demonstrating the benefits of using these drugs in combination (244).
Other Effects
WEIGHT LOSS
GLP-1 receptor agonists induce weight loss (16,239). A comparison of the ability of the maximum dose of different GLP-1 receptor analogues to induce weight loss are shown in table 23. It should be recognized that the weight loss shown in Table 23 represents averages. In clinical practice some patients lose a large amount of weight with GLP-1 receptor agonists while other patients can actually gain weight. The author has personally seen patients’ loss more than 50 lbs. Higher doses of liraglutide and semaglutide are approved for the treatment of obesity, which is discussed in the Endotext chapter “Pharmacologic Treatment of Overweight and Obesity in Adults” (245) (semagulutide 2.0mg is also approved for the treatment of diabetes). Studies have compared the effect of high doses of GLP-1 receptor agonists used for weight loss and lower doses used for treating diabetes (table 24). In general, higher doses of GLP-1 receptor agonists result in a modest further lowering of A1c and a more robust decrease in body weight.
|
Table 23. Effect of GLP-Receptor Agonists on Mean Weight Loss (12)
|
|
GLP-1 Receptor Agonist
|
Mean Weight Loss
|
|
Dulaglutide 1.5mg weekly
|
1.1Kg
|
|
Exenatide 10ug bid
|
1.2Kg
|
|
Exenatide 2mg weekly
|
1.1Kg
|
|
Liraglutide 1.8mg qd
|
1.5Kg
|
|
Lixisenatide 20ug qd
|
0.7Kg
|
|
Semaglutide 1mg weekly
|
3.8Kg
|
Based on a baseline weight of 90 kg after 26 weeks of treatment
|
Table 24. Comparison of Low and High Dose GLP-1 Receptor Agonists on A1c and Body Weight
|
|
|
Change in A1c (%)
|
Change in Body Weight (% or kg)
|
|
SCALE Diabetes (246)
|
|
Placebo
|
-0.3%
|
-2.0%
|
|
Liraglutide 1.8mg qd
|
-1.1%
|
-4.7%
|
|
Liraglutide 3.0mg qd
|
-1.3%
|
-6.0%
|
|
STEP-2 (247)
|
|
Placebo
|
-0.4%
|
-3.4%
|
|
Semaglutide 1mg weekly
|
-1.5%
|
-7.0%
|
|
Semaglutide 2.4mg weekly
|
-1.6%
|
-9.6%
|
|
SUSTAIN FORTE (248)
|
|
Semaglutide 1mg weekly
|
-1.9%
|
-6.2%
|
|
Semaglutide 2.0mg weekly
|
-2.2%
|
-7.2%
|
|
AWARD-11 (240)
|
|
|
|
Dulaglutide 1.5mg weekly
|
-1.5%
|
-3.1kg
|
|
Dulaglutide 3.0mg weekly
|
-1.7%
|
-4.0kg
|
|
Dulaglutide 4.5mg weekly
|
-1.9%
|
-4.7kg
|
The exact mechanisms responsible for the decrease in weight are not yet fully understood but both central and peripheral mechanisms are thought to play a part in activating receptors in the central nervous system associated with weight loss (239). GLP‐1 receptor agonists are thought to reduce body weight through decreased gastrointestinal motility and the promotion of satiety via the activation of GLP‐1 receptors in various regions of the brain (239).
BLOOD PRESSURE
GLP-1 receptor agonists result in modest but significant reductions in systolic blood pressure (2-5 mmHg) (16).
HEART RATE
The effects of GLP-1 receptor agonists on heart rate differ between drugs. Short-acting GLP-1 receptor agonists result in a modest increase (1-3 beats per minute) while long-acting GLP-1 receptor agonists are associated with a more pronounced and sustained increase (3-10 beats per minute) during the day and night (249).
LIPIDS
GLP-1 receptor agonists can favorably affect the lipid profile by inducing weight loss (decreasing triglycerides and very modestly decreasing LDL-C levels) (77). In a review by Nauck and colleagues it was noted that GLP-1 receptor agonists lowered TG levels by 18 to 62mg/dl depending upon the specific GLP-1 receptor agonist while decreasing LDL-C by 3-8mg/dl and increasing HDL-C by less than 1mg/dl (224). Additionally, GLP-1 receptor agonists reduce postprandial triglycerides by reducing circulating chylomicrons by decreasing intestinal lipoprotein production (77,224).
CARDIOVASCULAR DISEASE
The effect of six GLP-1 receptor agonists on cardiovascular disease has been reported.
ELIXA
In the Elixa trial 6,068 patients with T2DM and who recently had a myocardial infarction or been hospitalized for unstable angina were randomized to placebo or lixisenatide, and followed for a median of 25 months (250). The primary end point of cardiovascular death, myocardial infarction, stroke, or hospitalization for unstable angina was similar in the placebo or lixisenatide groups.
LEADER Trial
In contrast, the LEADER trial has shown that liraglutide decreased cardiovascular events (251). In this trial 9,340 patients with T2DM at high cardiovascular risk (~ 81% with established cardiovascular disease) were randomly assigned to receive liraglutide or placebo. After a median time of 3.5 years, the primary outcome of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke occurred in significantly fewer patients in the liraglutide group (13.0%) than in the placebo group (14.9%) (hazard ratio, 0.87, P=0.01). Additionally, deaths from cardiovascular causes (hazard ratio 0.78, P=0.007) or any cause was lower in the liraglutide group than in the placebo group (hazard ratio, 0.85; P=0.02). Interestingly patients with established cardiovascular disease or decreased renal function (eGFR < 60) appeared to derive the greatest benefit of liraglutide treatment (252,253). The decrease in cardiovascular events were similar in patients with and without a history of heart failure (254). Finally, a significant reduction in amputations with liraglutide vs. placebo was observed (HR 0.65; P = 0.03]) (255).
SUSTAIN 6 Trial
In support of the beneficial effects of some GLP1 receptor agonists to reduce cardiovascular events, semaglutide has also been shown to reduce cardiovascular events (256). In this trial, 3,297 patients with T2DM with established cardiovascular disease (83%), chronic heart failure, chronic kidney disease, or age >60 with at least one cardiovascular risk factor were randomized to receive once-weekly semaglutide (0.5 mg or 1.0 mg) or placebo for 104 weeks. The primary outcome of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke occurred in 6.6% of the semaglutide group and 8.9% of the placebo group (hazard ratio, 0.74; P = 0.02).
EXSCEL Trial
The effect of once weekly exenatide vs. placebo on cardiovascular outcomes was tested in 14,752 patients with T2DM, 73% who had cardiovascular disease (257). The primary outcome was the occurrence of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke. After a median follow-up of 3.2 years (duration of drug exposure 2.4 years) the primary outcome was reduced in the exenatide treated group but this difference just missed achieving statistical significance (hazard ratio 0.91; 95% CI 0.83-1.00; p=0.06). While not statistically significant these results are consistent with the results observed with other GLP-1 receptor agonists. It should be recognized that a high percentage of patients discontinued exenatide therapy in this trial (>40%) and this could have adversely affected the ability of exenatide treatment to favorably effect cardiovascular outcomes.
HARMONY Outcomes Trial
The effect of once weekly albiglutide vs. placebo was tested in 9,463 patients with T2DM and cardiovascular disease (258). The primary outcome was first occurrence of cardiovascular death, myocardial infarction, or stroke. After a median follow-up of 1.6 years a 22% decrease in the primary endpoint was observed in the albiglutide group (hazard ratio 0.78, p<0·0001). It should be noted that albiglutide is no longer available as it was removed from the market due to commercial considerations by the manufacturer.
REWIND Trial
This was a randomized study of weekly dulaglutide (1.5 mg) or placebo in 9,901 patients with T2DM who had either a previous cardiovascular event or cardiovascular risk factors (approximately 70% of patients did not have prior cardiovascular disease) (259). During a median follow-up of 5.4 years the primary outcome of non-fatal myocardial infarction, non-fatal stroke, or death from cardiovascular causes was decreased by 12% in the dulaglutide treated group (HR 0.88, p=0.026). The decrease in events was similar in participants with and without previous cardiovascular disease. In an analysis that focused on stroke it was noted that dulaglutide reduced ischemic stroke by 25% compared to placebo but had no effect on hemorrhagic stroke (260).
Summary
Thus, four studies have clearly demonstrated that treatment with GLP-1 receptor agonists reduces cardiovascular events, one study has provided data consistent with these results, and one study failed to demonstrate benefit. Why there are differences in results between these studies is unknown but could be due to differential effects of the GLP-1 receptor agonists, differences in the patient populations studied, or other unrecognized variables. A meta-analysis of 7 cardiovascular outcome studies using GLP-1 receptor agonists (ELIXA (lixisenatide), LEADER (liraglutide), SUSTAIN-6 (semaglutide), EXSCEL (exenatide), Harmony Outcomes (albiglutide), REWIND (dulaglutide), and PIONEER 6 (oral semaglutide) reported a 12% decrease in cardiovascular death, stroke, or myocardial infarction (p<0.0001), 12% decrease in cardiovascular deaths (p<0.003), 16% decrease in fatal or non-fatal strokes (p<0·0001), and 9% decrease in fatal or non-fatal myocardial infarctions (p=0.043) (261) (Table 25).
|
Table 25. Summary of GLP-1 Receptor Agonist Cardiovascular Outcome Trials
|
|
|
Number
|
Prior CVD
|
HbA1c
|
Mean Follow-up (years)
|
Hazard Ratio* (95% CI)
|
P value
|
|
ELIXA
Lixisenatide
|
6068
|
100%
|
7.7%
|
2.1
|
1.02
(0.89-1.17)
|
0.78
|
|
LEADER
Liraglutide
|
9340
|
81%
|
8.7%
|
3.8
|
0.87
(0.78-0.97)
|
0.015
|
|
SUSTAIN 6
Semaglutide
|
3297
|
83%
|
8.7%
|
2.1
|
0.74
(0.58-0.95)
|
0.016
|
|
EXSCEL
Exenatide
|
14,752
|
73%
|
8.0%
|
3.2
|
0.91
(0.83-1.00)
|
0.061
|
|
HARMONY
Albiglutide
|
9463
|
100%
|
8.7%
|
1.6
|
0.78
(0.68-0.90)
|
<0.001
|
|
REWIND
Dulaglutide
|
9901
|
31%
|
7.3%
|
5.4
|
0>88
(0.79-0.99)
|
0.026
|
|
PIONEER 6**
Semaglutide oral
|
3183
|
85%
|
8.2%
|
1.3
|
0.79
(0.57-1.11)
|
0.17
|
|
Overall (261)
|
|
|
|
|
0.88
(0.82-0.94)
|
<0.001
|
*CVD death, MI, Stroke. ** The Pioneer study is included in this table to provide information on all the studies examining the effect of GLP-1 receptor agonists on cardiovascular disease.
The mechanism accounting for this decrease in cardiovascular disease is uncertain but could be related to reductions in glycated hemoglobin, body weight, systolic blood pressure, postprandial triglyceride levels, or the direct effect of activation of GLP-1 receptors on the atherosclerotic process such as improving endothelial function (262).
The effect of a GLP-1 receptor agonist (efpeglenatide- not available) in patients on an SGLT-2 inhibitor was determined in the AMPLITUDE-O trial (263). The effect of efpeglenatide vs. placebo on cardiovascular and renal outcomes was similar in the absence and presence of baseline SGLT-2 inhibitors. Additional studies on the effect of the combination of GLP-1 agonists and SGLT-2 inhibitors on key outcomes are needed.
HEART FAILURE
Two small randomized studies have specifically examined the effect of GLP-1 receptor agonists on clinical outcomes in patients with heart failure. Margulies and colleagues randomized patients recently hospitalized for heart failure with a decreased ejection fraction to liraglutide (n=154) or placebo (n = 146) (59% with T2DM) (264). Treatment with liraglutide did not lead to greater posthospitalization clinical stability or decrease the number of deaths or rehospitalizations for heart failure. Jorsal et al carried out a randomized trial of liraglutide vs. placebo in patients (n=241) with reduced left ventricular ejection fraction who were clinically stable and on optimal heart failure treatment (265). Unexpectedly, serious cardiac events were seen in 10% of patients treated with liraglutide compared with 3% of patients in the placebo group (P = 0.04).
Several of the large cardiovascular outcome trials have analyzed the effect of administration of GLP-1 receptor agonists in the subgroup of patients with a history of heart failure. In the EXSCEL trial patients with heart failure at baseline had no decrease in all-cause mortality
whereas mortality was reduced in the subgroup without HF (HR 0.79; CI 0.68–0.92) (266). Similarly, in the combined data from the SUSTAIN-6 and PIONEER-6, patients with prior heart failure were the only subgroup that did not have a decrease in cardiovascular events (267). In contrast, in the LEADER trial the decrease in cardiovascular events were similar in patients with and without a history of heart failure (254).
The large cardiovascular outcome studies have determined the effect of GLP-1 receptor agonists on heart failure events. In a meta-analysis of the seven large cardiovascular outcome trials with a combined total of 56,004 participants, hospital admission for heart failure was decreased by 9% (0.91, 0.83-0.99; p=0.028) (261) (Table 26).
|
Table 26. Effect of GLP-1 Receptor Agonists on Heart Failure
|
|
Cardiovascular Outcome Trial
|
Heart Failure Hospitalization Heart Failure (HR (CI))
|
|
ELIXA (lixisenatide)
|
0.96 (0.75–1.23)
|
|
LEADER (liraglutide)
|
0.87 (0.73–1.05)
|
|
SUSTAIN-6 (semaglutide)
|
1.11 (0.77–1.61)
|
|
EXSCEL (exenatide)
|
0.94 (0.78–1.13)
|
|
HARMONY (albiglutide)
|
0.71 (0.53–0.94)
|
|
PIONEER-6 (oral semaglutide)
|
0.86 (0.48–1.55)
|
|
REWIND (dulaglutide)
|
0.93 (0.77–1.12)
|
|
Meta-analysis (261)
|
0.91 (0.83–0.99)
|
HR= hazard ratio; CI= 95% confidence interval
The effect of GLP-1 receptor agonists in preventing the development of heart failure and in patients with heart failure requires further study.
RENAL DISEASE
Five of the cardiovascular outcome studies described above also examined the effect of GLP-1 receptor agonists on kidney disease.
ELIXA Trial
Lixisenatide treatment decreased urinary albumin-to-creatinine ratio in patients with pre-existing micro or macroalbuminuria (268). Additionally, lixisenatide was associated with a reduced risk of new-onset macroalbuminuria compared with placebo (268). However, no significant differences in eGFR decline or the number of patients doubling their serum creatinine levels were seen between the lixisenatide treated group vs. placebo group (268).
LEADER Trial
The renal outcome in this trial was a composite of new-onset persistent macroalbuminuria, persistent doubling of the serum creatinine level, end-stage renal disease, or death due to renal disease. The renal outcome occurred in fewer patients in the liraglutide group than in the placebo group (hazard ratio, 0.78; P=0.003) (269). This favorable outcome was driven primarily by a decrease in the development of macroalbuminuria. The renal benefits did not appear to be driven by changes in A1c, body weight, or decreases in systolic BP.
SUSTAIN 6 Trial
In this trial, new or worsening nephropathy, defined as persistent macroalbuminuria, persistent doubling of the serum creatinine, or a creatinine clearance < 45ml/min/1.73m2, occurred in 3.8% of the patients in the semaglutide group and 6.1% of the patients in the placebo group (hazard ratio, 0.64; P=0.005) (256). As seen in the LEADER trial this favorable outcome was driven primarily by a decrease in the development of macroalbuminuria.
EXSCEL Trial
Exenatide treatment resulted in a reduction in new‐onset macroalbuminuria compared with placebo (2.2% vs 2.8%, P = 0.031), with no significant changes in either microalbuminuria (7.2% vs 7.5%) or ESKD requiring renal replacement therapy (0.7% vs 0.9%) (257).
REWIND Trial
The renal outcome included the occurrence of new macroalbuminuria (UACR >33·9 mg/mmol), a sustained decline in eGFR of 30% or more from baseline, or chronic renal replacement therapy (270). During a median follow-up of 5·4 years the renal outcome developed in 17.1% of patients in the dulaglutide group and in 19.6% of patients in the placebo group (HR 0.85, p=0·0004). This beneficial effect was driven by a reduction in the development of macroalbuminuria (HR 0.77; p<0.0001)
Summary
These studies demonstrate that GLP-1 receptor agonist administration reduce albuminuria without effecting eGFR. The decrease in albuminuria without effecting eGFR is similar to what was observed in some of the DPP-4 inhibitor studies described above. The mechanism accounting for this decrease is uncertain but decreased systolic BP, weight loss, improved glycemic control, or direct effects on the kidneys could have contributed to this decrease in albuminuria.
While the large studies described above demonstrated that GLP-1 receptor agonists primarily decrease albuminuria the AWARD 7 trial provides data on eGFR. The Award 7 was a multicenter randomized trial of dulaglutide 0.75mg weekly (n= 190), 1.5mg weekly (n= 193), or daily insulin glargine (n= 194) in patients with T2DM and Stage 3 and 4 chronic kidney disease (271). At 52 weeks, eGFR was higher with dulaglutide 1.5 mg (34.0; p=0.005 vs insulin glargine) and dulaglutide 0.75 mg (33.8; p=0·009 vs insulin glargine) than with insulin glargine (31.3mL/min per 1·73 m2). In contrast to the cardiovascular studies described above at 52 weeks dulaglutide 1.5 mg and 0.75 mg did not affect albuminuria. Additionally, a pooled analysis of the LEADER (liraglutide) and SUSTAIN 6 trials found a preservation in eGFR with GLP-1 receptor agonists, particularly in patients with a reduced baseline eGFR (272). Further studies are in progress to better define the effect of GLP1 receptor agonists on diabetic kidney disease.
NONALCOHOLIC FATTY LIVER DISEASE (NAFLD) AND NONALCOHOLIC STEATOHEPATITIS (NASH)
Studies have suggested that GLP-1 receptor agonists have beneficial effects on NAFLD and NASH (96). A meta-analysis of liraglutide studies and a separate meta-analysis of lixisenatide studies have reported that these drugs decrease liver enzymes (273,274). A 12-week randomized trial in 60 patients with NAFLD of exenatide + basal insulin vs. rapid acting insulin + basal insulin demonstrated lower liver enzymes in the exenatide treated group (275). Moreover, the reversal rate of fatty liver was greater in the group treated with exenatide (93.3%) than the intensive insulin group (66.7%) (p < 0.01). Similarly, liraglutide and dulaglutide has also been shown to decrease intrahepatic fat (276-278).
In the LEAN Trial 52 patients with NASH were randomized to liraglutide 1.8 mg daily or placebo and followed for 48 weeks (279). Resolution of NASH occurred in 39% of patients treated with liraglutide and only 9% patients in the placebo group (RR 4.3; p=0.019). Progression of fibrosis occurred in 9% of patients in the liraglutide group versus 36% patients in the placebo group (p=0.04).
A recent trial of semaglutide subcutaneously given daily (0.1, 0.2, and 0.4 mg) demonstrated an improvement in NASH without a beneficial effect on fibrosis (280). Whether weekly semaglutide or daily oral semaglutide would have similar effects is unknown.
While these data are suggestive larger and longer studies on the effect of GLP-1 receptor agonists on NAFLD and NASH are required.
Side Effects
GASTROINTESTINAL
The most common adverse effects are GI and include nausea, vomiting, and diarrhea (239). These symptoms are usually transient, resolving overtime (16). The GI side effects can be reduced by slowly increasing the dose (16). GI side effects tend to be more pronounced with short acting GLP-1 receptor agonists (239). Dehydration can occur secondary to GI side effects and can result in acute kidney failure (package insert).
GALL BLADDER DISEASE
Observational studies have shown an association of treatment with GLP-1 receptor agonists and bile duct and gallbladder disease (281). Additionally, a meta-analysis of randomized trials using GLP-1 inhibitors reported an association with an increased risk of cholelithiasis (282). Higher doses and a longer duration of treatment increased the risk of gallbladder disease (283). Finally, large cardiovascular trials with liraglutide (LEADER Trial), exenatide (EXSCEL Trial), and lixisenatide (ELIXA Trial) also reported an increased risk of gall bladder or biliary tract disease (250,257,284), however the large cardiovascular trial with semaglutide (SUSTAIN 6) did not observe an increase (256). It has been hypothesized that weight loss and/or decreased gallbladder motility induced by GLP-1 receptor agonists could contribute to this increase in gall bladder disease.
INJECTION-SITE REACTIONS
Injection-site reactions (rash, erythema) are also common with GLP-1 receptor agonists (16). Subcutaneous injection-site nodules may occur with the use of weekly exenatide (package insert), an abnormality that is due to the formulation.
MEDULLARY THYROID CANCER
Thyroid C-cell hyperplasia and medullary cell carcinoma has also been raised as possible concerns based on preclinical studies in rodents, but clinical studies in humans have not shown any indication of thyroid disorders (16). A meta-analysis of the four large cardiovascular outcome studies described above did not demonstrate an increased risk of medullary thyroid cancer with GLP-1 receptor agonist treatment (285)
PANCREATITIS
Subclinical increases in pancreatic enzyme levels are commonly observed with all GLP‐1 receptor agonists and pancreatitis has been reported (239). Importantly increases in lipase and amylase were not predictive of subsequent pancreatitis (286). A meta-analysis of four large cardiovascular outcome studies described above did not demonstrate an increased risk of pancreatitis or pancreatic cancer with GLP-1 receptor agonist treatment (285,287). A meta-analysis of all seven cardiovascular outcome studies also did not demonstrate an increase in pancreatitis with GLP-1 receptor agonist treatment (288).
RETINOPATHY
In the SUSTAIN 6 trial described above the rates of retinopathy complications (vitreous hemorrhage, blindness, or conditions requiring treatment with an intravitreal agent or photocoagulation) were significantly higher in the semaglutide group compared to the placebo group (hazard ratio, 1.76; P=0.02) (256). This increased risk of retinopathy complications has been attributed to the magnitude and rapidity of A1c reduction during the first 16 weeks of treatment in patients who had pre-existing retinopathy and poor glycemic control at baseline (“early worsening”) (289). A meta-analysis of GLP-1 cardiovascular trials found an association between retinopathy and the magnitude of A1c reduction supporting the hypothesis that the increase in retinopathy in SUSTAIN 6 was due to lowering of A1c (290).
Of note, other trials using semaglutide did not observe an increased risk of retinopathy (289). Additionally, an increase in diabetic retinopathy was not observed in the other cardiovascular outcome trials (250,251,257,258). In a meta-analysis of 60 studies with 60,077 patients treatment with GLP-1 receptor agonists did not increase the incidence of diabetic retinopathy, macular edema, retinal detachment, or retinal hemorrhage (291). However, the incidence of vitreous hemorrhage was higher in subjects treated with GLP-1 receptor agonists compared with placebo (odds ratios 1.93; 95% CI 1.09 to 3.42). Thus, it does not appear that GLP-1 receptor agonists treatment result in an increase in diabetic eye disease. A 5 years eye safety study for semaglutide, the FOCUS trial (NCT03811561), is currently underway
Contraindications and Drug Interactions
RENAL
Care needs to be exercised in patients with severe renal disease as they are more susceptible to the side effects of GLP-1 receptor agonists and more likely to have serious side effects (package inserts). There is limited data in patients with end stage renal disease.
Exenatide should not be used in patients with severe renal impairment (creatinine clearance < 30 mL/min) or end-stage renal disease (package insert). Caution should be applied when initiating or escalating doses of exenatide from 5 mcg to 10 mcg in patients with moderate renal impairment (creatinine clearance 30 to 50 mL/min) (package insert).
Weekly exenatide is not recommended for use in patients with eGFR below 45 mL/min/1.73m2 or end stage renal disease (package insert).
Lixisenatide is not recommended in patients with end stage renal disease (eGFR <15 mL/min/1.73 m2) (package insert).
No dose adjustments for liraglutide, semaglutide, or dulaglutide are recommended for patients with renal impairment (package insert).
OTHER
Exenatide is not recommended in patients with gastroparesis or severe gastrointestinal disease (package insert).
In patients with a history of pancreatitis or at high risk for pancreatitis many clinicians avoid GLP-1 receptor agonists.
GLP-1 receptor agonists are contraindicated in patients with a personal or family history of Medullary Thyroid Cancer and in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2) (package insert).
Summary
The ability of GLP-1 receptor agonists to effectively decrease A1c levels, reduce atherosclerotic cardiovascular disease, and in some patients induce a major loss of weight make these drugs very attractive in the treatment of patients with T2DM. Additionally, once weekly administration for certain drugs in this class can improve compliance.
|
Table 27. Advantages and Disadvantages of GLP- 1 Receptor Agonists
|
|
Advantages
|
Disadvantages
|
|
Weight Loss
|
GI side effects
|
|
No Hypoglycemia
|
Requires Injection
|
|
Reduce CVD (liraglutide, semaglutide, dulaglutide)
|
Pancreatitis?
|
|
Improve NAFLD
|
Thyroid cancer?
|
|
Once a week therapy possible
|
Gall bladder disease
|
|
Decrease albuminuria
|
Expensive
|
|
Decrease postprandial glucose
|
|
ORAL GLUCAGON LIKE PROTEIN-1 (GLP-1) RECEPTOR AGONISTS
Introduction
In 2019 an oral form of semaglutide became available. To facilitate absorption of semaglutide, which is a 31 amino acid peptide, the tablet contains a permeation enhancer N-(8-[2-hydroxybenzoyl]amino)caprylic acid (SNAC, Eligen® Technology, Emisphere Technologies), which is a small fatty acid derivative that accelerates the absorption of semaglutide across the gastric epithelium avoiding the activation of proteolytic enzymes and pH-induced degradation in the stomach (292). This allows for the absorption of an intact peptide. One should note that the bioavailability of oral semaglutide is very low as the dose of oral semaglutide is 7-14 mg per day vs 0.5-2.0 mg once a week with the injectable dose.
Administration
The oral form of semaglutide must be taken at least 30 minutes before the first food, beverage, or other oral medications of the day with no more than 4 ounces of plain water (package insert). Waiting less than 30 minutes, or taking with food, beverages (other than plain water), or other oral medications will adversely affect the absorption of semaglutide. Waiting more than 30 minutes to eat may increase the absorption. The starting dose is 3 mg once daily for 30 days. After 30 days on the 3 mg dose, increase the dose to 7 mg once daily. The dose may be increased to 14 mg once daily if additional glycemic control is needed after at least 30 days on the 7 mg dose (package insert). Patients treated with once weekly semaglutide 0.5 mg injections can be transitioned to oral semaglutide 7 mg or 14 mg a day. No dose adjustment is recommended for patients with renal or hepatic impairment (package insert).
Mechanism of Action
The mechanism of action is identical to injected GLP-1 receptor agonists described above.
Glycemic Efficacy
In a meta-analysis of five trials of oral semaglutide vs. placebo, treatment with oral semaglutide reduced HbA1c by 0.89% (293). In the Pioneer 1 study 703 patients were randomized (mean baseline HbA1c 8.0%) to placebo vs. various doses of oral semaglutide (294). After 26 weeks of treatment A1c decreased by -0.6% in the 3 mg group, -0.9% in the 7 mg group, and -1.1% in the 14 mg group compared to placebo (P < 0.001 for all results). If the decrease in A1c was adjusted for premature drug discontinuation or initiation of rescue medication the estimated decreases in A1c were -0.7% in the 3 mg group, -1.2% in the 7 mg group, and -1.4% in the 14 mg group (P < 0.001 for all).
Studies have also examined the ability of oral semaglutide to lower A1c vs. other drugs. Compared to sitagliptin, oral semaglutide 7mg per day reduced A1c by -0.3% while 14mg per day reduced A1c by 0.5% (P < .001 for both) (295). In a similar trial with flexible dose adjustment of semaglutide, treatment with semaglutide (60% on 14mg per day) resulted in a 1.4% decrease in A1c while 100mg sitagliptin decreased A1c by 0.7% (296). In a trial comparing empagliflozin vs. oral semaglutide, treatment with semaglutide resulted in a greater decrease in A1c compared to empagliflozin (-1.3% vs. -0.9%; P < 0.0001) (297). In a comparison of liraglutide 1.8mg per day vs. oral semaglutide 14mg per day the change from baseline in A1c was -1.2% (SE 0·1) with oral semaglutide and -1.1% with subcutaneous liraglutide (298). If the decrease in A1c was adjusted for premature drug discontinuation or initiation of rescue medication then oral semaglutide treatment resulted in a slightly greater decreases in A1c than subcutaneous liraglutide (estimated treatment difference -0·2%). Finally, early in the development of oral semaglutide various doses of oral semaglutide were compared to weekly injected semaglutide (299). Compared to placebo 10mg per day of oral semaglutide reduced A1c by –1.2%, 20mg by –1.4%, while 1mg per week of injected semaglutide decreased A1c by 1.9% (not significantly different than the 20mg oral dose). Thus, oral semaglutide is more effective in lowering A1c levels than DPP-4 inhibitors or SGLT2 inhibitors and similar to liraglutide and perhaps slightly less potent than injected semaglutide.
Other Effects
WEIGHT LOSS
In a meta-analysis of weight loss, treatment with oral semaglutide reduced body weight by 2.99 kg compared to placebo (293). In a 26-week study comparing sitagliptin vs. oral semaglutide the 7mg dose resulted in a 1.6kg decrease and the 14mg dose a 2.5kg decrease in weight compared to sitagliptin (295). In contrast, oral semaglutide 14mg and empagliflozin 25mg resulted in a similar decrease in body weight at 26-weeks (-3.8 vs. -3.7kg) and 52-weeks (-3.8 vs. -3.6kg) (297). Finally, in a 26-week trial oral semaglutide resulted in greater weight loss (-4.4 kg than liraglutide (-3·1 kg) (298).
BLOOD PRESSURE AND PULSE RATE
In a meta-analysis of blood pressure, treatment with oral semaglutide reduced systolic blood pressure by 3.16 mmHg and increased pulse rate by 1.90 beats per minute compared with placebo (293).
CARDIOVASCULAR DISEASE
3,183 patients with T2DM at high cardiovascular risk (age of ≥50 years with established cardiovascular or chronic kidney disease, or age of ≥60 years with cardiovascular risk factors) were randomly assigned to receive oral semaglutide or placebo (300). After a median time of 15.9 months, major adverse cardiovascular events, the primary outcome, occurred in 3.8% of the subjects treated with oral semaglutide and 4.8% of the placebo group (HR 0.79; 95% CI 0.57 to 1.11). Deaths from cardiovascular causes were 0.9% in the oral semaglutide group and 1.9% in the placebo group (HR 0.49; 95% CI, 0.27 to 0.92) while death from any cause occurred in 1.4% in the oral semaglutide group and 2.8% in the placebo group (HR 0.51; 95% CI, 0.31 to 0.84). It should be noted that the primary outcome was not statistically decreased in this study, which may be due to the relatively small number of subjects studied and the short duration of the study that together resulted in a small number of events. Additionally, more patients in the placebo group received treatment with an SGLT2 inhibitor than in the oral semaglutide group and SGLT2 inhibitors are well recognized to reduce cardiovascular disease events (see section on SGLT2 inhibitors), which could also have diminished the ability to observe a decrease in events in the oral semaglutide group. Because the glucose lowering, weight loss, and many other effects of oral semaglutide are very similar to injected semaglutide many experts consider the effects on cardiovascular and renal disease to also be similar.
Side Effects
The most common adverse effects are GI and include nausea, vomiting, and diarrhea (292). Transient mild or moderate nausea was the most common adverse event occurring in 5-21% of subjects treated with oral semaglutide (292).
Severe hypoglycemia is uncommon in patients treated with oral semaglutide (292). The risk of hypoglycemia is increased when oral semaglutide is used in combination with insulin secretagogues (e.g., sulfonylureas) or insulin. Patients may require a lower dose of the secretagogue or insulin to reduce the risk of hypoglycemia when used in combination with oral semaglutide.
The safety profile of oral semaglutide is similar to other GLP-1 receptor agonists (see side effect section for GLP1 receptor agonists).
Contraindications and Drug Interactions
Similar to other GLP1 receptor agonists oral semaglutide is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2.
No notable drug interactions have been described (package insert).
Summary
The delivery of a GLP1 receptor agonist via the oral route is advantageous and make oral semaglutide an attractive choice in the treatment of patients with T2DM who do want to inject medications given its ability to decrease A1c, body weight, and blood pressure with few serious side effects. It is likely that the other beneficial effects of GLP1 receptor agonists (e.g., reducing cardiovascular disease and proteinuria) will also occur with the oral formulation.
DUEL GLP-1 RECEPTOR AND GIP RECEPTOR AGONIST
Introduction
Tirzepatide (Mounjaro) is a 39 amino acid peptide that was engineered from the native GIP sequence and has agonist activity at both the GIP and GLP-1 receptors (301,302). A C20 fatty diacid moiety is conjugated at the position 20 lysine residue, which facilitates binding to albumin thereby resulting a half-life after administration of approximately 5 days allowing for weekly administration (301,302).
Administration
Tirzepatide is administered weekly at any time of day, with or without meals. The starting dose is 2.5mg subcutaneously and after 4 weeks the dose is increased to 5 mg (302). Depending upon the response one may increase the dosage in 2.5 mg increments every 4 weeks to a maximum dose of 15 mg per week (302). No dosage adjustment is recommended for renal or hepatic disease (package insert).
Mechanism of Action
Both GLP-1 and GIP stimulate insulin secretion in a glucose dependent fashion (303). The higher the glucose the greater the effect with no effect when glucose levels are in the normal to low range (303). As one would expect tirzepatide stimulates both first- and second-phase insulin secretion (302,304). GLP-1 inhibits glucagon secretion when glucose levels are increased while GIP will stimulate glucagon secretion, particularly when glucose levels are in the normal to low range (303). Tirzepatide reduces fasting and postprandial glucagon concentrations (304). These effects on insulin and glucagon secretion lead to decreases in glucose levels with a low risk of hypoglycemia as the increase in insulin secretion and decrease in glucagon secretion are dependent on elevated glucose levels. In addition, tirzepatide improves insulin sensitivity (304,305). While this increase in insulin sensitivity may be due to weight loss studies suggest that there may be additional factors contributing to the improved insulin sensitivity (305). GIP may have peripheral effects that could enhance insulin sensitivity.
Pharmacologic levels of GLP-1 slow gastric emptying and induce satiety by activating receptors in the hypothalamus thereby leading to decreased food intake and weight loss (303). GIP also appears to have central effects leading to decreased food intake in rodents but the effect in humans is not well defined (303).
Glycemic Efficacy
A number of different studies (SURPASS trials) have examined the effect of 5mg, 10mg, and 15mg of tirzepatide on glycemic control under a variety of clinical situations (Table 28). SURPASS 1 compared tirzepatide vs. placebo in patients on no medications (306), SURPASS 2 compared tirzepatide vs. semaglutide at a dose of 1 mg in patients on metformin (307), SURPASS 3 compared tirzepatide vs. degludec insulin in patients on metformin alone or in combination with an SGLT2 inhibitor (308), SURPASS 4 compared tirzepatide vs glargine insulin in patients treated with any combination of metformin, sulfonylurea, or SGLT-2 inhibitor (309), and SURPASS 5 compared tirzepatide vs. placebo in patients treated with glargine insulin with or without metformin (310). The treatment duration was 40 weeks in SURPASS 1, 2, and 5 and 52 weeks in SURPASS 3 and 4. Baseline A1c levels were between 7.9% and 8.5% in the SURPASS studies.
|
Table 28. Decrease in HbA1c with Tirzepatide Treatment
|
|
|
SURPASS 1
|
SURPASS 2
|
SURPASS 3
|
SURPASS 4
|
SURPASS 5
|
|
|
Tirzepatide vs. Placebo
|
Tirzepatide vs. Semaglutide
|
Tirzepatide vs. Degludec
|
Tirzepatide vs.
Glargine
|
Tirzepatide vs. Placebo
|
|
Baseline A1c
|
7.9%
|
8.3%
|
8.2%
|
8.5%
|
8.3%
|
|
Tirzepatide 5mg
|
-1.8
|
-2.0
|
-1.9
|
-2.1
|
-2.1
|
|
Tirzepatide 10mg
|
-1.7
|
-2.2
|
-2.0
|
-2.3
|
-2.4
|
|
Tirzepatide 15mg
|
-1.7
|
-2.3
|
-2.1
|
-2.4
|
-2.3
|
|
Comparator
|
-0.1
|
-1.9
|
-1.3
|
-1.4
|
-0.9
|
It should be noted that the reduction in A1c induced by tirzepatide is quite impressive and results in an A1c level in an “intensive” control range. For example, in the SURPASS 2 trial 80% of patients had an A1c < 6.5% and 46% < 5.7% on 15mg tirzepatide. Additionally, comparison with semaglutide (SURPASS 2) demonstrated a modestly greater lowering of A1c with tirzepatide. A greater difference in the ability to decrease A1c was seen in an earlier study comparing tirzepatide vs. dulaglutide (tirzepatide 5mg- 1.6%, 10mg- 2.0%,15 mg- 2.4%; duluglutide 1.5mg- 1.1%) (311). Note the comparisons with semaglutide and dulaglutide used in these studies were not the maximal dose. Comparisons with insulin therapy (SURPASS 3 and 4) show better glycemic control with tirzepatide, which is likely due to an increased risk of hypoglycemia with insulin therapy that limits treatment. In SURPASS 3, 48% of patients on insulin therapy had a blood glucose < 70mg/dL while on tirzepatide treatment 8-14% of patients had a blood glucose < 70mg/dL. Severe hypoglycemia is not frequently observed with tirzepatide in the absence of concomitant insulin or sulfonylurea therapy. Finally, it is worth noting that the additional A1c reduction with an increased dose of tirzepatide is very modest. This is important to recognize that in patients that have side effects with higher doses of tirzepatide treatment it is not necessary to achieve maximal doses of tirzepatide to robustly improve glycemic control.
Other Effects
WEIGHT LOSS
Significant weight loss has been observed with tirzepatide administration. Table 29 shows the weight loss observed in the SURPASS trials. In contrast to the modest effects of increased doses of tirzepatide on A1c levels increased doses of tirzepatide have a greater effect on weight loss. At the 15mg dose over a 10% loss in weight is observed. It should be noted that in SURPASS 2 tirzepatide is compared to semaglutide 1.0mg, which is not the dose that is recommended for weight loss (the recommended dose is 2.4mg) and therefore one cannot be certain that tirzepatide is more efficacious than higher doses of semaglutide. In a comparison of tirzepatide vs. dulaglutide, tirzepatide resulted in greater weight loss (tirzepatide 5mg- 4.8kg, 10mg- 8.7kg, 15mg-11.3kg; dulaglutide 1.5mg- 2.7kg) (311).
|
Table 29. Decrease in Weight with Tirzepatide Treatment
|
|
|
SURPASS 1
|
SURPASS 2
|
SURPASS 3
|
SURPASS 4
|
SURPASS 5
|
|
|
Tirzepatide vs. Placebo
|
Tirzepatide vs. Semaglutide
|
Tirzepatide vs. Degludec
|
Tirzepatide vs.
Glargine
|
Tirzepatide vs. Placebo
|
|
Tirzepatide 5mg
|
-6.3kg/ -7.9%
|
-7.6kg/ -8.5%
|
-7.0kg/ -8.1%
|
-6.4kg/ -8.1%
|
-5.4kg/ -6.6%
|
|
Tirzepatide 10mg
|
-7.0kg/ -9.3%
|
-9.3kg/ -11.0%
|
-9.6kg/ -11.4%
|
-8.9kg/ -10.7%
|
-7.5kg/ -8.9%
|
|
Tirzepatide 15mg
|
-7.8kg/ -11.0%
|
-11.2kg/ -13.1%
|
-11.3kg/ -13.9%
|
-10.6kg/ -13.0%
|
-8.8kg/ -11.6%
|
|
Comparator
|
-1.0kg/ -0.9%
|
-5.7kg/ -6.7%
|
+1.9kg/ +2.7%
|
+1.7kg/ +2.2%
|
+1.6kg/ +1.7%
|
BLOOD PRESSURE AND PULSE
In the SURPASS studies described above tirzepatide treatment decreased systolic BP by 2.8 to 12.6 mm Hg and diastolic BP by 0.8 to 4.5 mm Hg (301). Tirzepatide treatment increased heart rate by approximately 2 to 4 beats per minute.
LIPIDS
In the SURPASS studies described above plasma triglyceride levels were consistently decreased by 13-25% (table 30). In most studies with the exception of SURPASS 5, HDL cholesterol levels increased by 3-11%. Total cholesterol and LDL cholesterol levels modestly decreased in most studies. Not unexpectedly given the decrease in triglyceride levels small LDL particles were decreased (312). The decrease in triglycerides could be related to weight loss, which is well known to affect these parameters (313). Additionally, GIP and tirzepatide increase lipoprotein lipase activity, which could increase the clearance of triglyceride rich lipoproteins (303,312). Finally, tirzepatide lowered Apo-CIII levels, which could also play a role in the decrease in triglyceride levels (312).
|
Table 30. Effect of Tirzapetide 15mg on Lipid Levels
|
|
|
SURPASS 1
|
SURPASS 2
|
SURPASS 3
|
SURPASS 4
|
SURPASS 5
|
|
|
Tirzepatide vs. Placebo
|
Tirzepatide vs. Semaglutide
|
Tirzepatide vs. Degludec
|
Tirzepatide vs.
Glargine
|
Tirzepatide vs. Placebo
|
|
Total Cholesterol
|
-7.6%
|
-1.5%
|
-3.0%
|
-5.6%
|
-12.6%
|
|
Triglycerides
|
-25.7%
|
-13.3%
|
-13.0%
|
-16.1%
|
-19.4%
|
|
LDLc
|
-10.8%
|
+1.2%
|
-3.8%
|
-9.3%
|
-17.3%
|
|
HDLc
|
+11.3%
|
+2.7%
|
+9.2%
|
+7.9%
|
-0.8%
|
Results are percent change in tirzepatide group minus percent change in comparator group
CARDIOVASCULAR DISEASE
A meta-analysis of seven randomized controlled trials with 4,887 participants treated with tirzepatide and 2,328 control participants found that MACE 4 (cardiovascular death, myocardial infarction, stroke, and hospitalized unstable angina) was decreased but not statistically significant (HR 0.80; 95% CI, 0.57–1.11) (314). One should note that the number of events in this meta-analysis was small because the duration of these studies was relatively short (approximately 1 year) and the population of patients included in these studies were not at high risk for cardiovascular events (only 1/3 with pre-existing cardiovascular disease). A long-term trial dedicated to determining the effect of tirzepatide on cardiovascular disease is ongoing (SURPASS-CVOT trial NCT04255433).
LIVER DISEASE
Liver fat content was decreased to a greater degree with tirzepatide treatment compared to treatment with insulin degludec (315). Additionally, tirzepatide decreased alanine aminotransferase and aspartate aminotransferase levels (316). Further studies on the effect of tirzepatide on liver disease are in progress.
Side Effects
The side effects described in the section on GLP-1 receptor agonists also are of concern with tirzepatide.
Patients treated with tirzepatide in combination with a sulfonylurea or insulin may have an increased risk of hypoglycemia. The risk of hypoglycemia may be decreased by a reduction in sulfonylurea or insulin dose.
The incidence of pancreatitis was increased in patients treated with tirzepatide compared to comparator treatment ((0.23 patients per 100 years of exposure vs. 0.11 patients per 100 years of exposure) (package insert). Additionally, acute gallbladder disease (cholelithiasis, biliary colic, and cholecystectomy) was increased with tirzepatide treatment (0.6% of tirzepatide-treated patients and 0% of placebo-treated patients) (package insert).
As with other GLP-1 receptor agonists nausea, diarrhea, vomiting, dyspepsia, constipation, and decreased appetite are common side effects.
Contraindications and Drug Interactions
Tirzepatide is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with MEN2. Tirzepatide has not been studied in patients with a prior history of pancreatitis and it is unknown if patients with a history of pancreatitis are at higher risk for developing pancreatitis.
Tirzepatide delays gastric emptying and thereby has the potential to impact the absorption of concomitantly administered oral medications. The delay is largest after the first dose and diminishes over time.
SUMMARY
The major advantage of tirzepatide compared to GLP-1 receptor agonists is the greater decrease in weight and A1c levels.
INSULIN-GLP-1 RECEPTOR AGONIST COMBINATIONS
Introduction
There are currently two insulin-GLP-1 receptor agonist combinations available for use; glargine insulin/lixisenatide (iGlarLixi) (Soliqua) and degludec insulin/liraglutide (iDegLira) (Xultophy). Both combine a basal insulin with a once-a-day GLP-1 receptor agonist. iGlarLixi contains 100U glargine and 33 ug lixisenatide per ml. iDegLira contains 100U degludec insulin and 3.6 mg liraglutide per ml.
Administration
In patients naive to basal insulin or to a GLP-1 receptor agonist, currently on a GLP-1 receptor agonist, or currently on less than 30 units of basal insulin daily the recommended starting dosage of iGlarLixi 100/33 is 15 units (15 units insulin glargine/5 ug lixisenatide) given subcutaneously once daily. In patients currently on 30 to 60 units of basal insulin daily, with or without a GLP-1 receptor agonist the recommended starting dosage of iGalLixi 100/33 is 30 units (30 units insulin glargine/10 ug lixisenatide) given subcutaneously once daily. After starting with the recommended dose, titrate the dosage upwards or downwards by two to four units weekly based on the patient’s glycemic control until the desired fasting plasma glucose is achieved. Administer iGlarLixi 100/33 subcutaneously once a day within an hour prior to the first meal of the day. The maximum dose of iGlarLixi 100/33 is 60 units daily (60 units insulin glargine/20 ug lixisenatide).
In patients naive to basal insulin or GLP-1 receptor agonist therapy the recommended starting dose of iDegLira 100/3.6 is 10 units (10 units of insulin degludec and 0.36 mg of liraglutide) given subcutaneously once-daily. In patients currently on basal insulin or a GLP-1 receptor agonist the recommended starting dose of iDegLira 100/3.6 is 16 units (16 units of insulin degludec and 0.58 mg of liraglutide) given subcutaneously once-daily. After starting the recommended starting dose, titrate the dosage upwards or downwards by two units every three to four days based on the patient’s blood glucose monitoring results and glycemic control goal until the desired fasting plasma glucose is achieved. Administer iDegLira 100/3.6 by subcutaneous injection once-daily at the same time each day with or without food. The maximum dose of iDegLira 100/3.6 is 50 units daily (50 units of insulin degludec and 1.8 mg of liraglutide).
Mechanism of Action
Basal insulin regulates fasting blood glucose levels between meals and overnight while a GLP-1 receptor agonist lowers postprandial glucose levels (317). Together this drug combination results in 24-hour glycemic control.
Glycemic Efficacy
A number of studies have compared the ability of the combination of insulin-GLP receptor agonists to lower A1c levels compared to either insulin alone or GLP-1 receptor agonist alone (317). Table 31 shows the results of two large studies. As shown in Table 31 combination therapy was better at lowering A1c levels compared to the individual components (317). Additionally, the risk of hypoglycemia was similar with combination therapy compared to basal insulin alone. In a study of patients poorly controlled on glargine insulin adding rapid acting insulin (basal/bolus therapy) vs. switching to iDegLira was found to result in a similar reduction in A1c levels but the risk of hypoglycemia was greater with basal/bolus insulin (318). Not unexpectedly basal/bolus insulin resulted in greater weight gain (difference 3.6 kg) (318). Indirect comparisons suggest that iDegLira reduces A1C slightly more (< 0.5%) than iGlarLixi but this could be due to different study design, different patient populations, or other differences between the trials (317). A meta-analysis of 8 studies concluded that iDegLira and iGlarLixi demonstrated no significant differences in absolute HbA1c changes, fasting plasma glucose levels, or body weight changes relative to baseline (319).
|
Table 31. Effect of Combination Therapy vs Individual Components on Key Outcomes
|
|
Study
|
Treatment
|
A1c Reduction
|
% Subjects with Hypoglycemia
|
Change in Body Weight (Kg)
|
|
Rosenstock et al (320)
|
iGlarLixi
|
1.6%
|
26
|
-0.3
|
|
|
Glar
|
1.3%
|
24
|
+1.1
|
|
|
Lixi
|
0.9%
|
6
|
-2.3
|
|
Gough et al (321)
|
iDegLira
|
1.9%
|
32
|
-0.5
|
|
|
Deg
|
1.4%
|
39
|
+1.6
|
|
|
Lira
|
1.3%
|
7
|
-3.0
|
Other Effects
As shown in Table 31 the typical weight gain seen with insulin therapy alone is blunted with combination therapy.
Side Effects
Studies have noted that the typical GI side effects seen with GLP-1 receptor agonist therapy is blunted with combination therapy (141). The likely explanation is that the titration of the GLP-1 receptor agonist is slower with combination therapy (141).
Contraindications
The maximum daily insulin dose of 60 units for iGlarLixi and 50 units for iDegLira, may not be sufficient in patients requiring higher daily basal insulin doses (e.g., patients with severe insulin resistance). The maximum dose is determined by the GLP-1 receptor agonist dose (the max dose of iDegLira delivers 1.8 mg of liraglutide while the max dose of iGlarLixa delivers 20 ug of lixisenatide). Conversely, there may be some patients who require only a low dose of basal insulin and thus because of the fixed ratio of basal insulin to GLP-1 receptor agonist the dose of the GLP-1 receptor agonist may be too low. These examples are a limitation of fixed ratio delivery systems. In these patients one can use basal insulin and a GLP-1 receptor agonist independently. It should be noted that for the majority of patients the fixed ratio will be acceptable.
Summary
The effects of combination therapy are predictable based on studies of basal insulin and GLP-1 receptor agonists but providing them in a single injection provides convenience and makes it easier for patients to comply. Additionally, these combination drugs are titrated based on fasting glucose values and therefore frequent home blood glucose monitoring is not required, which also makes compliance easier. In patients who do not have adequate control on basal insulin alone combination therapy can be a useful therapeutic option.
BILE ACID SEQUESTRANTS
Introduction
Colesevelam (Welchol) is a non-absorbed, polymeric, LDL cholesterol lowering and glucose lowering agent that is a high-capacity bile acid-binding molecule (322). This drug was developed primarily to lower LDL cholesterol levels and was later noted to have favorable effects on blood glucose levels and was approved for improving glycemic control in patients with T2DM (16,322). It should be noted that other bile acid sequestrants (cholestyramine) also have favorable effects on glycemic control (323).
Administration
The recommended dose of colesevelam is 6 tablets once daily or 3 tablets twice daily with meals (tabs 625 mg). Alternatively, one can take one 3.75-gram packet once daily mixed with water, fruit juice, or diet soft drinks and taken with meals or one flavored chewable bar (80 calories per bar) with meals. For patients with difficulty swallowing tablets the use of packets or chewable bars is recommended.
Mechanism of Action
The mechanism by which bile acid sequestrants improve glucose metabolism is not well understood and the literature on this topic is often contradictory (324,325). Colesevelam does not alter hepatic or peripheral insulin sensitivity or decrease glucose GI absorption (325,326). Neither acute nor chronic treatment affect post oral glucose tolerance test blood glucose levels (326). Additionally, colesevelam treatment did not alter endogenous glucose production, gluconeogenesis, or glycogenolysis (325,326). Thus, the mechanisms accounting for the decrease in glucose effect of bile acid sequestrants remain unclear.
A leading hypothesis is that bile acid sequestrants improve glucose metabolism by stimulating the incretin pathway. Colesevelam increases GLP-1 and GIP concentrations suggesting that an increase in incretins contributes to the improvement in glycemic control (326-328). There are two pathways by which colesevelam increases GLP-1 secretion; (1) TGR5-mediated GLP-1 secretion in L cells and (2) intestinal proglucagon expression.
TGR5 is a G protein–coupled receptor expressed in many organs and tissues including the intestine (326,328). Bile acids activate TGR5 on the surface of intestinal L cells promoting GLP-1 secretion (326,328,329). Bile acid sequestrants appear to augment TGR5 activation and GLP-1 release, which occurs primarily in the distal intestine and colon (326,329,330).
FXR is a nuclear hormone receptor that complexes with RXR to alter the expression of a large number of genes (328). Bile acids are a ligand for FXR and activate FXR thereby regulating gene expression (328). FXR activation in intestinal L cells decreases proglucagon expression resulting in a decrease in GLP-1 production (331). Conversely, a decrease in bile acids due to binding to colesevelam increases GLP-1 gene expression and secretion in response to glucose improving glucose metabolism (331).
It is likely that there are both incretin dependent and independent mechanisms that account for the improvement in glycemic control with colesevelam treatment. The exact mechanisms by which bile acid sequestrants lower A1c levels remain to be elucidated.
Glycemic Efficacy
Colesevelam has modest effects on glycemic control, lowering A1c levels by approximately 0.5% when added to metformin, sulfonylureas, pioglitazone, or insulin (16,322,332). Colesevelam does not lead to an increase in weight (322). In combination with metformin hypoglycemia is not a problem but when used in combination with insulin or sulfonylureas hypoglycemia may occur (322).
Other Effects
LIPIDS
Colesevelam lowers LDL cholesterol levels by 15-20% and has only a modest effect on HDL cholesterol levels (322,333). The effect of bile acid sequestrants on triglyceride levels varies (333). In patients with normal triglyceride levels, bile acid sequestrants increase triglyceride levels by a small amount. However, as baseline triglyceride levels increase, the effect of bile acid sequestrants on plasma triglyceride levels becomes greater, and can result in substantial increases in triglyceride levels (333). In patients with triglycerides > 500mg/dl the use of bile acid sequestrants is contraindicated (333).
CARDIOVASCULAR DISEASE
There have been no randomized studies that have examined the effect of bile acid sequestrants on cardiovascular end points in subjects with diabetes. In non-diabetic-subjects bile acid sequestrants have reduced cardiovascular events (334,335). Since bile acid sequestrants have a similar beneficial impact on serum lipid levels in diabetic and non-diabetic subjects one would anticipate that these drugs would also result in a reduction in events in the diabetic population.
Side Effects
Colesevelam does not have major systemic side effects as it is not absorbed and remains in the intestinal tract (333). However, it does cause gastrointestinal (GI) side effects (333). Constipation is a common side effect and can be severe. In addition, patients will often complain of bloating, dyspepsia, abdominal discomfort, and aggravation of hemorrhoids. Because of GI distress, a small number of patients will discontinue therapy with colesevelam. One can reduce or ameliorate these GI side effects by increasing hydration, adding fiber to the diet (psyllium), and using stool softeners.
Contraindications and Drug Interactions
Colesevelam treatment is contraindicated in patients with a history of bowel obstruction and is cautioned in those with a history of gastrointestinal motility disorders (i.e., gastroparesis) or gastrointestinal surgery (322,333). Colesevelam is contraindicated in patients with plasma triglyceride levels > 500 mg/dL or a history of hypertriglyceridemia-induced pancreatitis (package insert).
In the intestine bile acid sequestrants can impede the absorption of many other drugs (333). Colesevelam, which requires a much lower quantity of drug because of its high affinity and binding capacity for bile salts, has less of an effect on the absorption of other drugs than other bile acid sequestrants but can still adversely affect the absorption of certain drugs (Table 32) (333). Administration of these drugs, as well as vitamin supplements, 4 hours prior to administration of colesevelam minimizes pharmacokinetic interactions (333). This is particularly important with drugs that have a narrow toxic/therapeutic window, such as thyroid hormone, digoxin, or warfarin. It can be difficult for some patients, particularly those on multiple medications, to take colesevelam given the need to separate pill ingestion.
|
Table 32. Drugs Affected by Colesevelam
|
|
L-thyroxine
|
Cyclosporine
|
Glimepiride
|
Glipizide
|
|
Glyburide
|
Phenytoin
|
Olmesartan
|
Warfarin
|
|
Oral contraceptives
|
Repaglinide
|
Fenofibrate
|
Vitamin Supplements
|
Colesevelam may also decrease the absorption of fat-soluble vitamins A, D, E, and K (package insert).
Summary
Colesevelam has the advantage of lowering both A1c and LDL cholesterol levels. However, the efficacy of lowering A1c and LDL cholesterol levels is modest compared to other drugs. Additionally, in our patients with diabetes who are often on multiple medications it can be difficult to coordinate taking colesevelam with these other medications.
|
Table 33. Advantages and Disadvantages of Colesevelam
|
|
Advantages
|
Disadvantages
|
|
Lowers LDL cholesterol
|
Increases triglyceride levels particularly if already high
|
|
Minimal systemic effects
|
GI side effects
|
|
Once a day administration possible
|
Inhibits the absorption of other drugs
|
|
No hypoglycemia
|
Modest effect on A1c
|
|
Weight neutral
|
Relatively Expensive
|
PRAMLINTIDE (SYMLIN)
Introduction
Pramlintide is a soluble synthetic analog of human amylin (336). Amylin is co-sequestered and co-secreted with insulin by the pancreatic beta cells in response to nutrient stimuli (336). Amylin secretion in response to nutrients is absent in type 1 diabetes and in patients with T2DM there is impaired beta-cell secretion of amylin in response to nutrients (336). Amylin suppresses post-prandial arginine-stimulated glucagon secretion, suppresses appetite, and slows gastric emptying time through effects on the brain (336).
Administration
In patients with T2DM initiate pramlintide at 60 ug subcutaneously immediately prior to each major meal. Increase the dose from 60 to 120 ug prior to each major meal when no clinically significant nausea has occurred for at least 3 days. Note the dose used to treat patients with Type 1 diabetes differs from the dose used in patients with T2DM.
Mechanism of Action
Pramlintide attenuates post-prandial glucagon secretion, enhances satiety, and reduces food intake, which together improve glycemic control (336). These effects are mediated centrally (336)
Glycemic Efficacy
In a review of three randomized trials in patients with T2DM comparing pramlintide vs. placebo the A1c level was decreased by approximately 0.3-0.6% in the pramlintide group (337). Postprandial glucose excursions are significantly blunted with the addition of pramlintide (336). Pramlintide has only minimal effects on fasting glucose levels (337).
In a study comparing rapid acting insulin vs. pramlintide with meals a similar reduction in A1c was observed (338). In contrast to rapid acting insulin, patients treated with pramlintide did not gain weight (338). Additionally, the frequency of hypoglycemia was less with pramlintide compared with rapid acting insulin (338).
Other Effects
Pramlintide treatment decreases weight (approximately 1-3 kg), which is likely due to decreased food intake (336,337). In a comparison of food intake during an ad libitum buffet meal, treatment with pramlintide resulted in an approximately 200 calorie decrease in food intake compared to placebo administration (339). Pramlintide also decreases gastric emptying (336).
Side Effects
A major side effect of pramlintide is nausea which can lead to patients discontinuing this drug (337).
Although pramlintide alone does not cause hypoglycemia, in combination with rapid acting meal time insulin the two drugs synergistically increase the risk of severe hypoglycemia (336). Therefore, rapid acting meal time insulin needs to be reduced upon initiation of pramlintide treatment to decrease the risk of hypoglycemia (336). Reducing rapid acting meal time insulin by 30-50% is recommended during the initial dose titration period (336).
Contraindications and Drug Interactions
Pramlintide is contraindicated in patients with hypoglycemia unawareness and confirmed gastroparesis (package insert).
Summary
Pramlintide is currently seldom used. Its modest effect on A1c levels coupled with the difficulties of administration (extra injections) and side effects has led to minimal use of this agent. Additionally, its major advantage of weight loss is now superseded by the use of GLP-1 receptor agonists.
|
Table 34. Advantages and Disadvantages of Pramlintide
|
|
Advantages
|
Disadvantages
|
|
Weight loss
|
Hypoglycemia
|
|
Decrease postprandial glucose
|
Frequent dosing
|
|
|
GI side effects
|
|
|
Expensive
|
|
|
Modest reduction in A1c
|
SUMMARY
A large number of drugs are now available for lowering glucose levels. For information on the management of T2DM and selecting amongst the available pharmacological agents see the chapter by Schroeder (5). For information on the use of these drugs to treat diabetes during pregnancy, in children and adolescents, and for the prevention of diabetes see other Endotext chapters (2-4).
REFERENCES
- American Diabetes Association. 5. Lifestyle Management: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019; 42:S46-S60
- Buschur E, Stetson B, Barbour LA. Diabetes In Pregnancy. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2018.
- Perreault L. Prediabetes. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2022.
- Yau M, Sperling M. Treatment of Diabetes mellitus in Children and Adolescents. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2021.
- Schroeder EB. Management of Type 2 Diabetes: Selecting Amongst Available Pharmacological Agents. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2022.
- Davies MJ, D'Alessio DA, Fradkin J, Kernan WN, Mathieu C, Mingrone G, Rossing P, Tsapas A, Wexler DJ, Buse JB. Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care2018; 41:2669-2701
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020; 43:S98-S110
- Buse JB, Wexler DJ, Tsapas A, Rossing P, Mingrone G, Mathieu C, D'Alessio DA, Davies MJ. 2019 Update to: Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2020; 43:487-493
- Cersosimo E, Triplitt C, Solis-Herrera C, Mandarino LJ, DeFronzo RA. Pathogenesis of Type 2 Diabetes Mellitus. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2021.
- Bloomgarden ZT, Dodis R, Viscoli CM, Holmboe ES, Inzucchi SE. Lower baseline glycemia reduces apparent oral agent glucose-lowering efficacy: a meta-regression analysis. Diabetes Care 2006; 29:2137-2139
- DeFronzo RA, Stonehouse AH, Han J, Wintle ME. Relationship of baseline HbA1c and efficacy of current glucose-lowering therapies: a meta-analysis of randomized clinical trials. Diabet Med 2010; 27:309-317
- Maloney A, Rosenstock J, Fonseca V. A Model-Based Meta-Analysis of 24 Antihyperglycemic Drugs for Type 2 Diabetes: Comparison of Treatment Effects at Therapeutic Doses. Clin Pharmacol Ther 2019; 105:1213-1223
- Nathan DM, Buse JB, Kahn SE, Krause-Steinrauf H, Larkin ME, Staten M, Wexler D, Lachin JM. Rationale and design of the glycemia reduction approaches in diabetes: a comparative effectiveness study (GRADE). Diabetes Care 2013; 36:2254-2261
- Thule PM, Umpierrez G. Sulfonylureas: a new look at old therapy. Curr Diab Rep 2014; 14:473
- Khunti K, Chatterjee S, Gerstein HC, Zoungas S, Davies MJ. Do sulphonylureas still have a place in clinical practice? Lancet Diabetes Endocrinol 2018; 6:821-832
- Tahrani AA, Barnett AH, Bailey CJ. Pharmacology and therapeutic implications of current drugs for type 2 diabetes mellitus. Nat Rev Endocrinol 2016; 12:566-592
- Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab 2013; 18:162-185
- Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015; 58:221-232
- MacDonald PE, Wheeler MB. Voltage-dependent K(+) channels in pancreatic beta cells: role, regulation and potential as therapeutic targets. Diabetologia 2003; 46:1046-1062
- Ashcroft FM, Gribble FM. ATP-sensitive K+ channels and insulin secretion: their role in health and disease. Diabetologia 1999; 42:903-919
- Zhang CL, Katoh M, Shibasaki T, Minami K, Sunaga Y, Takahashi H, Yokoi N, Iwasaki M, Miki T, Seino S. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 2009; 325:607-610
- DeFronzo RA. Pharmacologic therapy for type 2 diabetes mellitus. Ann Intern Med 1999; 131:281-303
- Sherifali D, Nerenberg K, Pullenayegum E, Cheng JE, Gerstein HC. The effect of oral antidiabetic agents on A1C levels: a systematic review and meta-analysis. Diabetes Care 2010; 33:1859-1864
- Phung OJ, Scholle JM, Talwar M, Coleman CI. Effect of noninsulin antidiabetic drugs added to metformin therapy on glycemic control, weight gain, and hypoglycemia in type 2 diabetes. JAMA 2010; 303:1410-1418
- Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O'Neill MC, Zinman B, Viberti G. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med2006; 355:2427-2443
- Goldner MG, Knatterud GL, Prout TE. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. 3. Clinical implications of UGDP results. JAMA 1971; 218:1400-1410
- Meinert CL, Knatterud GL, Prout TE, Klimt CR. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. II. Mortality results. Diabetes 1970; 19:Suppl:789-830
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:837-853
- Rosenstock J, Kahn SE, Johansen OE, Zinman B, Espeland MA, Woerle HJ, Pfarr E, Keller A, Mattheus M, Baanstra D, Meinicke T, George JT, von Eynatten M, McGuire DK, Marx N. Effect of Linagliptin vs Glimepiride on Major Adverse Cardiovascular Outcomes in Patients With Type 2 Diabetes: The CAROLINA Randomized Clinical Trial. JAMA 2019;
- Silbert R, Salcido-Montenegro A, Rodriguez-Gutierrez R, Katabi A, McCoy RG. Hypoglycemia Among Patients with Type 2 Diabetes: Epidemiology, Risk Factors, and Prevention Strategies. Curr Diab Rep 2018; 18:53
- Middleton TL, Wong J, Molyneaux L, Brooks BA, Yue DK, Twigg SM, Wu T. Cardiac Effects of Sulfonylurea-Related Hypoglycemia. Diabetes Care 2017; 40:663-670
- Hay LC, Wilmshurst EG, Fulcher G. Unrecognized hypo- and hyperglycemia in well-controlled patients with type 2 diabetes mellitus: the results of continuous glucose monitoring. Diabetes Technol Ther 2003; 5:19-26
- Gangji AS, Cukierman T, Gerstein HC, Goldsmith CH, Clase CM. A systematic review and meta-analysis of hypoglycemia and cardiovascular events: a comparison of glyburide with other secretagogues and with insulin. Diabetes Care 2007; 30:389-394
- Tuttle KR, Bakris GL, Bilous RW, Chiang JL, de Boer IH, Goldstein-Fuchs J, Hirsch IB, Kalantar-Zadeh K, Narva AS, Navaneethan SD, Neumiller JJ, Patel UD, Ratner RE, Whaley-Connell AT, Molitch ME. Diabetic kidney disease: a report from an ADA Consensus Conference. Diabetes Care 2014; 37:2864-2883
- By the American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2019 Updated AGS Beers Criteria(R) for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc2019; 67:674-694
- Guardado-Mendoza R, Prioletta A, Jimenez-Ceja LM, Sosale A, Folli F. The role of nateglinide and repaglinide, derivatives of meglitinide, in the treatment of type 2 diabetes mellitus. Arch Med Sci 2013; 9:936-943
- Tran L, Zielinski A, Roach AH, Jende JA, Householder AM, Cole EE, Atway SA, Amornyard M, Accursi ML, Shieh SW, Thompson EE. Pharmacologic treatment of type 2 diabetes: oral medications. Ann Pharmacother2015; 49:540-556
- Scott LJ. Repaglinide: a review of its use in type 2 diabetes mellitus. Drugs 2012; 72:249-272
- Horton ES, Clinkingbeard C, Gatlin M, Foley J, Mallows S, Shen S. Nateglinide alone and in combination with metformin improves glycemic control by reducing mealtime glucose levels in type 2 diabetes. Diabetes Care2000; 23:1660-1665
- Rosenstock J, Hassman DR, Madder RD, Brazinsky SA, Farrell J, Khutoryansky N, Hale PM. Repaglinide versus nateglinide monotherapy: a randomized, multicenter study. Diabetes Care 2004; 27:1265-1270
- Navigator Study Group, Holman RR, Haffner SM, McMurray JJ, Bethel MA, Holzhauer B, Hua TA, Belenkov Y, Boolell M, Buse JB, Buckley BM, Chacra AR, Chiang FT, Charbonnel B, Chow CC, Davies MJ, Deedwania P, Diem P, Einhorn D, Fonseca V, Fulcher GR, Gaciong Z, Gaztambide S, Giles T, Horton E, Ilkova H, Jenssen T, Kahn SE, Krum H, Laakso M, Leiter LA, Levitt NS, Mareev V, Martinez F, Masson C, Mazzone T, Meaney E, Nesto R, Pan C, Prager R, Raptis SA, Rutten GE, Sandstroem H, Schaper F, Scheen A, Schmitz O, Sinay I, Soska V, Stender S, Tamas G, Tognoni G, Tuomilehto J, Villamil AS, Vozar J, Califf RM. Effect of nateglinide on the incidence of diabetes and cardiovascular events. N Engl J Med 2010; 362:1463-1476
- Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia 2017; 60:1577-1585
- Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab 2014; 20:953-966
- LaMoia TE, Shulman GI. Cellular and Molecular Mechanisms of Metformin Action. Endocr Rev 2021; 42:77-96
- Buse JB, DeFronzo RA, Rosenstock J, Kim T, Burns C, Skare S, Baron A, Fineman M. The Primary Glucose-Lowering Effect of Metformin Resides in the Gut, Not the Circulation: Results From Short-term Pharmacokinetic and 12-Week Dose-Ranging Studies. Diabetes Care 2016; 39:198-205
- Pollak M. The effects of metformin on gut microbiota and the immune system as research frontiers. Diabetologia 2017; 60:1662-1667
- Sanchez-Rangel E, Inzucchi SE. Metformin: clinical use in type 2 diabetes. Diabetologia 2017; 60:1586-1593
- Williams LK, Padhukasahasram B, Ahmedani BK, Peterson EL, Wells KE, Gonzalez Burchard E, Lanfear DE. Differing effects of metformin on glycemic control by race-ethnicity. J Clin Endocrinol Metab 2014; 99:3160-3168
- Fujioka K, Pans M, Joyal S. Glycemic control in patients with type 2 diabetes mellitus switched from twice-daily immediate-release metformin to a once-daily extended-release formulation. Clin Ther 2003; 25:515-529
- Wulffele MG, Kooy A, de Zeeuw D, Stehouwer CD, Gansevoort RT. The effect of metformin on blood pressure, plasma cholesterol and triglycerides in type 2 diabetes mellitus: a systematic review. J Intern Med 2004; 256:1-14
- Feingold KR. Role of Glucose and Lipids in the Cardiovascular Disease of Patients with Diabetes. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2020.
- Ratner R, Goldberg R, Haffner S, Marcovina S, Orchard T, Fowler S, Temprosa M. Impact of intensive lifestyle and metformin therapy on cardiovascular disease risk factors in the diabetes prevention program. Diabetes Care2005; 28:888-894
- Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:854-865
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577-1589
- Kooy A, de Jager J, Lehert P, Bets D, Wulffele MG, Donker AJ, Stehouwer CD. Long-term effects of metformin on metabolism and microvascular and macrovascular disease in patients with type 2 diabetes mellitus. Arch Intern Med 2009; 169:616-625
- Hong J, Zhang Y, Lai S, Lv A, Su Q, Dong Y, Zhou Z, Tang W, Zhao J, Cui L, Zou D, Wang D, Li H, Liu C, Wu G, Shen J, Zhu D, Wang W, Shen W, Ning G. Effects of metformin versus glipizide on cardiovascular outcomes in patients with type 2 diabetes and coronary artery disease. Diabetes Care 2013; 36:1304-1311
- Legro RS. Evaluation and Treatment of Polycystic Ovary Syndrome. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2017.
- Heckman-Stoddard BM, DeCensi A, Sahasrabuddhe VV, Ford LG. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 2017; 60:1639-1647
- Mallik R, Chowdhury TA. Metformin in cancer. Diabetes Res Clin Pract 2018; 143:409-419
- Goodwin PJ, Chen BE, Gelmon KA, Whelan TJ, Ennis M, Lemieux J, Ligibel JA, Hershman DL, Mayer IA, Hobday TJ, Bliss JM, Rastogi P, Rabaglio-Poretti M, Mukherjee SD, Mackey JR, Abramson VG, Oja C, Wesolowski R, Thompson AM, Rea DW, Stos PM, Shepherd LE, Stambolic V, Parulekar WR. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients With Breast Cancer: The MA.32 Randomized Clinical Trial. JAMA 2022; 327:1963-1973
- Dujic T, Zhou K, Donnelly LA, Tavendale R, Palmer CN, Pearson ER. Association of Organic Cation Transporter 1 With Intolerance to Metformin in Type 2 Diabetes: A GoDARTS Study. Diabetes 2015; 64:1786-1793
- Dawed AY, Zhou K, van Leeuwen N, Mahajan A, Robertson N, Koivula R, Elders PJM, Rauh SP, Jones AG, Holl RW, Stingl JC, Franks PW, McCarthy MI, t Hart LM, Pearson ER. Variation in the Plasma Membrane Monoamine Transporter (PMAT) (Encoded by SLC29A4) and Organic Cation Transporter 1 (OCT1) (Encoded by SLC22A1) and Gastrointestinal Intolerance to Metformin in Type 2 Diabetes: An IMI DIRECT Study. Diabetes Care 2019; 42:1027-1033
- de Jager J, Kooy A, Lehert P, Wulffele MG, van der Kolk J, Bets D, Verburg J, Donker AJ, Stehouwer CD. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ 2010; 340:c2181
- Aroda VR, Edelstein SL, Goldberg RB, Knowler WC, Marcovina SM, Orchard TJ, Bray GA, Schade DS, Temprosa MG, White NH, Crandall JP. Long-term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab 2016; 101:1754-1761
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019; 42:S90-S102
- Yki-Jarvinen H. Thiazolidinediones. N Engl J Med 2004; 351:1106-1118
- Willson TM, Lambert MH, Kliewer SA. Peroxisome proliferator-activated receptor gamma and metabolic disease. Annu Rev Biochem 2001; 70:341-367
- Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C, Feingold KR. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 2000; 141:4021-4031
- Aronoff S, Rosenblatt S, Braithwaite S, Egan JW, Mathisen AL, Schneider RL. Pioglitazone hydrochloride monotherapy improves glycemic control in the treatment of patients with type 2 diabetes: a 6-month randomized placebo-controlled dose-response study. The Pioglitazone 001 Study Group. Diabetes Care 2000; 23:1605-1611
- Dennis JM, Shields BM, Henley WE, Jones AG, Hattersley AT. Disease progression and treatment response in data-driven subgroups of type 2 diabetes compared with models based on simple clinical features: an analysis using clinical trial data. Lancet Diabetes Endocrinol 2019; 7:442-451
- Tan MH, Baksi A, Krahulec B, Kubalski P, Stankiewicz A, Urquhart R, Edwards G, Johns D. Comparison of pioglitazone and gliclazide in sustaining glycemic control over 2 years in patients with type 2 diabetes. Diabetes Care 2005; 28:544-550
- Gastaldelli A, Ferrannini E, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. Thiazolidinediones improve beta-cell function in type 2 diabetic patients. Am J Physiol Endocrinol Metab 2007; 292:E871-883
- Xiang AH, Peters RK, Kjos SL, Marroquin A, Goico J, Ochoa C, Kawakubo M, Buchanan TA. Effect of pioglitazone on pancreatic beta-cell function and diabetes risk in Hispanic women with prior gestational diabetes. Diabetes 2006; 55:517-522
- DeFronzo RA, Inzucchi S, Abdul-Ghani M, Nissen SE. Pioglitazone: The forgotten, cost-effective cardioprotective drug for type 2 diabetes. Diab Vasc Dis Res 2019; 16:133-143
- Sarafidis PA, Stafylas PC, Georgianos PI, Saratzis AN, Lasaridis AN. Effect of thiazolidinediones on albuminuria and proteinuria in diabetes: a meta-analysis. Am J Kidney Dis 2010; 55:835-847
- Qayyum R, Adomaityte J. A meta-analysis of the effect of thiazolidinediones on blood pressure. J Clin Hypertens (Greenwich) 2006; 8:19-28
- Ferrannini E, DeFronzo RA. Impact of glucose-lowering drugs on cardiovascular disease in type 2 diabetes. Eur Heart J 2015; 36:2288-2296
- Spanheimer R, Betteridge DJ, Tan MH, Ferrannini E, Charbonnel B. Long-term lipid effects of pioglitazone by baseline anti-hyperglycemia medication therapy and statin use from the PROactive experience (PROactive 14). Am J Cardiol 2009; 104:234-239
- Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 2011; 364:127-135
- Deeg MA, Buse JB, Goldberg RB, Kendall DM, Zagar AJ, Jacober SJ, Khan MA, Perez AT, Tan MH. Pioglitazone and rosiglitazone have different effects on serum lipoprotein particle concentrations and sizes in patients with type 2 diabetes and dyslipidemia. Diabetes Care 2007; 30:2458-2464
- Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, Tan MH, Khan MA, Perez AT, Jacober SJ. A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care 2005; 28:1547-1554
- Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, Skene AM, Tan MH, Lefebvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L, Betteridge J, Birkeland K, Golay A, Heine RJ, Koranyi L, Laakso M, Mokan M, Norkus A, Pirags V, Podar T, Scheen A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J, Smith U, Taton J. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005; 366:1279-1289
- Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, Guarino PD, Lovejoy AM, Peduzzi PN, Conwit R, Brass LM, Schwartz GG, Adams HP, Jr., Berger L, Carolei A, Clark W, Coull B, Ford GA, Kleindorfer D, O'Leary JR, Parsons MW, Ringleb P, Sen S, Spence JD, Tanne D, Wang D, Winder TR. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N Engl J Med 2016; 374:1321-1331
- Spence JD, Viscoli CM, Inzucchi SE, Dearborn-Tomazos J, Ford GA, Gorman M, Furie KL, Lovejoy AM, Young LH, Kernan WN. Pioglitazone Therapy in Patients With Stroke and Prediabetes: A Post Hoc Analysis of the IRIS Randomized Clinical Trial. JAMA Neurol 2019; 76:526-535
- Vaccaro O, Masulli M, Nicolucci A, Bonora E, Del Prato S, Maggioni AP, Rivellese AA, Squatrito S, Giorda CB, Sesti G, Mocarelli P, Lucisano G, Sacco M, Signorini S, Cappellini F, Perriello G, Babini AC, Lapolla A, Gregori G, Giordano C, Corsi L, Buzzetti R, Clemente G, Di Cianni G, Iannarelli R, Cordera R, La Macchia O, Zamboni C, Scaranna C, Boemi M, Iovine C, Lauro D, Leotta S, Dall'Aglio E, Cannarsa E, Tonutti L, Pugliese G, Bossi AC, Anichini R, Dotta F, Di Benedetto A, Citro G, Antenucci D, Ricci L, Giorgino F, Santini C, Gnasso A, De Cosmo S, Zavaroni D, Vedovato M, Consoli A, Calabrese M, di Bartolo P, Fornengo P, Riccardi G. Effects on the incidence of cardiovascular events of the addition of pioglitazone versus sulfonylureas in patients with type 2 diabetes inadequately controlled with metformin (TOSCA.IT): a randomised, multicentre trial. Lancet Diabetes Endocrinol 2017; 5:887-897
- Vaccaro O, Lucisano G, Masulli M, Bonora E, Del Prato S, Rivellese AA, Giorda CB, Mocarelli P, Squatrito S, Maggioni AP, Riccardi G, Nicolucci A. Cardiovascular Effects of Pioglitazone or Sulfonylureas According to Pretreatment Risk: Moving Toward Personalized Care. J Clin Endocrinol Metab 2019; 104:3296-3302
- Langenfeld MR, Forst T, Hohberg C, Kann P, Lubben G, Konrad T, Fullert SD, Sachara C, Pfutzner A. Pioglitazone decreases carotid intima-media thickness independently of glycemic control in patients with type 2 diabetes mellitus: results from a controlled randomized study. Circulation 2005; 111:2525-2531
- Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D'Agostino RB, Sr., Perez A, Provost JC, Haffner SM. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: a randomized trial. JAMA 2006; 296:2572-2581
- Saremi A, Schwenke DC, Buchanan TA, Hodis HN, Mack WJ, Banerji M, Bray GA, Clement SC, Henry RR, Kitabchi AE, Mudaliar S, Ratner RE, Stentz FB, Musi N, Tripathy D, DeFronzo RA, Reaven PD. Pioglitazone slows progression of atherosclerosis in prediabetes independent of changes in cardiovascular risk factors. Arterioscler Thromb Vasc Biol 2013; 33:393-399
- Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A, Jure H, De Larochelliere R, Staniloae CS, Mavromatis K, Saw J, Hu B, Lincoff AM, Tuzcu EM. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA2008; 299:1561-1573
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007; 356:2457-2471
- Singh S, Loke YK, Furberg CD. Long-term risk of cardiovascular events with rosiglitazone: a meta-analysis. JAMA 2007; 298:1189-1195
- Home PD, Pocock SJ, Beck-Nielsen H, Curtis PS, Gomis R, Hanefeld M, Jones NP, Komajda M, McMurray JJ. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet 2009; 373:2125-2135
- Mahaffey KW, Hafley G, Dickerson S, Burns S, Tourt-Uhlig S, White J, Newby LK, Komajda M, McMurray J, Bigelow R, Home PD, Lopes RD. Results of a reevaluation of cardiovascular outcomes in the RECORD trial. Am Heart J 2013; 166:240-249 e241
- Bach RG, Brooks MM, Lombardero M, Genuth S, Donner TW, Garber A, Kennedy L, Monrad ES, Pop-Busui R, Kelsey SF, Frye RL. Rosiglitazone and outcomes for patients with diabetes mellitus and coronary artery disease in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Circulation 2013; 128:785-794
- Kim KS, Lee BW, Kim YJ, Lee DH, Cha BS, Park CY. Nonalcoholic Fatty Liver Disease and Diabetes: Part II: Treatment. Diabetes Metab J 2019; 43:127-143
- Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, Berria R, Ma JZ, Dwivedi S, Havranek R, Fincke C, DeFronzo R, Bannayan GA, Schenker S, Cusi K. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006; 355:2297-2307
- Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, Tio F, Hardies J, Darland C, Musi N, Webb A, Portillo-Sanchez P. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann Intern Med 2016; 165:305-315
- Musso G, Cassader M, Paschetta E, Gambino R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern Med 2017; 177:633-640
- Rosenstock J, Vico M, Wei L, Salsali A, List JF. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA(1c), body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care 2012; 35:1473-1478
- Abdul-Ghani MA, Puckett C, Triplitt C, Maggs D, Adams J, Cersosimo E, DeFronzo RA. Initial combination therapy with metformin, pioglitazone and exenatide is more effective than sequential add-on therapy in subjects with new-onset diabetes. Results from the Efficacy and Durability of Initial Combination Therapy for Type 2 Diabetes (EDICT): a randomized trial. Diabetes Obes Metab 2015; 17:268-275
- Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton ES, Le Winter M, Porte D, Semenkovich CF, Smith S, Young LH, Kahn R. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256-263
- Mudaliar S, Chang AR, Henry RR. Thiazolidinediones, peripheral edema, and type 2 diabetes: incidence, pathophysiology, and clinical implications. Endocr Pract 2003; 9:406-416
- Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y, Breyer MD. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat Med 2005; 11:861-866
- Zanchi A, Chiolero A, Maillard M, Nussberger J, Brunner HR, Burnier M. Effects of the peroxisomal proliferator-activated receptor-gamma agonist pioglitazone on renal and hormonal responses to salt in healthy men. J Clin Endocrinol Metab 2004; 89:1140-1145
- de Jong M, van der Worp HB, van der Graaf Y, Visseren FLJ, Westerink J. Pioglitazone and the secondary prevention of cardiovascular disease. A meta-analysis of randomized-controlled trials. Cardiovasc Diabetol2017; 16:134
- Hernandez AV, Usmani A, Rajamanickam A, Moheet A. Thiazolidinediones and risk of heart failure in patients with or at high risk of type 2 diabetes mellitus: a meta-analysis and meta-regression analysis of placebo-controlled randomized clinical trials. Am J Cardiovasc Drugs 2011; 11:115-128
- Masoudi FA, Inzucchi SE, Wang Y, Havranek EP, Foody JM, Krumholz HM. Thiazolidinediones, metformin, and outcomes in older patients with diabetes and heart failure: an observational study. Circulation 2005; 111:583-590
- Kahn SE, Zinman B, Lachin JM, Haffner SM, Herman WH, Holman RR, Kravitz BG, Yu D, Heise MA, Aftring RP, Viberti G. Rosiglitazone-associated fractures in type 2 diabetes: an Analysis from A Diabetes Outcome Progression Trial (ADOPT). Diabetes Care 2008; 31:845-851
- Dormandy J, Bhattacharya M, van Troostenburg de Bruyn AR. Safety and tolerability of pioglitazone in high-risk patients with type 2 diabetes: an overview of data from PROactive. Drug Saf 2009; 32:187-202
- Viscoli CM, Inzucchi SE, Young LH, Insogna KL, Conwit R, Furie KL, Gorman M, Kelly MA, Lovejoy AM, Kernan WN. Pioglitazone and Risk for Bone Fracture: Safety Data From a Randomized Clinical Trial. J Clin Endocrinol Metab 2017; 102:914-922
- Zhu ZN, Jiang YF, Ding T. Risk of fracture with thiazolidinediones: an updated meta-analysis of randomized clinical trials. Bone 2014; 68:115-123
- Schwartz AV, Chen H, Ambrosius WT, Sood A, Josse RG, Bonds DE, Schnall AM, Vittinghoff E, Bauer DC, Banerji MA, Cohen RM, Hamilton BP, Isakova T, Sellmeyer DE, Simmons DL, Shibli-Rahhal A, Williamson JD, Margolis KL. Effects of TZD Use and Discontinuation on Fracture Rates in ACCORD Bone Study. J Clin Endocrinol Metab 2015; 100:4059-4066
- Schwartz AV, Sellmeyer DE, Vittinghoff E, Palermo L, Lecka-Czernik B, Feingold KR, Strotmeyer ES, Resnick HE, Carbone L, Beamer BA, Park SW, Lane NE, Harris TB, Cummings SR. Thiazolidinedione use and bone loss in older diabetic adults. J Clin Endocrinol Metab 2006; 91:3349-3354
- Davidson MB. Pioglitazone (Actos) and bladder cancer: Legal system triumphs over the evidence. J Diabetes Complications 2016; 30:981-985
- Erdmann E, Harding S, Lam H, Perez A. Ten-year observational follow-up of PROactive: a randomized cardiovascular outcomes trial evaluating pioglitazone in type 2 diabetes. Diabetes Obes Metab 2016; 18:266-273
- Lewis JD, Habel LA, Quesenberry CP, Strom BL, Peng T, Hedderson MM, Ehrlich SF, Mamtani R, Bilker W, Vaughn DJ, Nessel L, Van Den Eeden SK, Ferrara A. Pioglitazone Use and Risk of Bladder Cancer and Other Common Cancers in Persons With Diabetes. JAMA 2015; 314:265-277
- Levin D, Bell S, Sund R, Hartikainen SA, Tuomilehto J, Pukkala E, Keskimaki I, Badrick E, Renehan AG, Buchan IE, Bowker SL, Minhas-Sandhu JK, Zafari Z, Marra C, Johnson JA, Stricker BH, Uitterlinden AG, Hofman A, Ruiter R, de Keyser CE, MacDonald TM, Wild SH, McKeigue PM, Colhoun HM. Pioglitazone and bladder cancer risk: a multipopulation pooled, cumulative exposure analysis. Diabetologia 2015; 58:493-504
- Idris I, Warren G, Donnelly R. Association between thiazolidinedione treatment and risk of macular edema among patients with type 2 diabetes. Arch Intern Med 2012; 172:1005-1011
- Ryan EH, Jr., Han DP, Ramsay RC, Cantrill HL, Bennett SR, Dev S, Williams DF. Diabetic macular edema associated with glitazone use. Retina 2006; 26:562-570
- Coniff R, Krol A. Acarbose: a review of US clinical experience. Clin Ther 1997; 19:16-26; discussion 12-13
- Van de Laar FA, Lucassen PL, Akkermans RP, Van de Lisdonk EH, Rutten GE, Van Weel C. Alpha-glucosidase inhibitors for type 2 diabetes mellitus. Cochrane Database Syst Rev 2005:CD003639
- Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: the STOP-NIDDM trial. JAMA 2003; 290:486-494
- Yun P, Du AM, Chen XJ, Liu JC, Xiao H. Effect of Acarbose on Long-Term Prognosis in Acute Coronary Syndromes Patients with Newly Diagnosed Impaired Glucose Tolerance. J Diabetes Res 2016; 2016:1602083
- Holman RR, Coleman RL, Chan JCN, Chiasson JL, Feng H, Ge J, Gerstein HC, Gray R, Huo Y, Lang Z, McMurray JJ, Ryden L, Schroder S, Sun Y, Theodorakis MJ, Tendera M, Tucker L, Tuomilehto J, Wei Y, Yang W, Wang D, Hu D, Pan C. Effects of acarbose on cardiovascular and diabetes outcomes in patients with coronary heart disease and impaired glucose tolerance (ACE): a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2017; 5:877-886
- Domecq JP, Prutsky G, Leppin A, Sonbol MB, Altayar O, Undavalli C, Wang Z, Elraiyah T, Brito JP, Mauck KF, Lababidi MH, Prokop LJ, Asi N, Wei J, Fidahussein S, Montori VM, Murad MH. Clinical review: Drugs commonly associated with weight change: a systematic review and meta-analysis. J Clin Endocrinol Metab 2015; 100:363-370
- Lupsa BC, Inzucchi SE. Use of SGLT2 inhibitors in type 2 diabetes: weighing the risks and benefits. Diabetologia 2018; 61:2118-2125
- Thomas MC, Cherney DZI. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018; 61:2098-2107
- Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005; 54:3427-3434
- Monami M, Liistro F, Scatena A, Nreu B, Mannucci E. Short and medium-term efficacy of sodium glucose co-transporter-2 (SGLT-2) inhibitors: A meta-analysis of randomized clinical trials. Diabetes Obes Metab 2018; 20:1213-1222
- Ferrannini G, Hach T, Crowe S, Sanghvi A, Hall KD, Ferrannini E. Energy Balance After Sodium-Glucose Cotransporter 2 Inhibition. Diabetes Care 2015; 38:1730-1735
- Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med 2015; 373:2117-2128
- Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med 2017; 377:644-657
- Mazidi M, Rezaie P, Gao HK, Kengne AP. Effect of Sodium-Glucose Cotransport-2 Inhibitors on Blood Pressure in People With Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of 43 Randomized Control Trials With 22 528 Patients. J Am Heart Assoc 2017; 6
- Sanchez-Garcia A, Simental-Mendia M, Millan-Alanis JM, Simental-Mendia LE. Effect of sodium-glucose co-transporter 2 inhibitors on lipid profile: A systematic review and meta-analysis of 48 randomized controlled trials. Pharmacol Res 2020; 160:105068
- van Baar MJB, van Ruiten CC, Muskiet MHA, van Bloemendaal L, RG IJ, van Raalte DH. SGLT2 Inhibitors in Combination Therapy: From Mechanisms to Clinical Considerations in Type 2 Diabetes Management. Diabetes Care 2018; 41:1543-1556
- Chung MC, Hung PH, Hsiao PJ, Wu LY, Chang CH, Wu MJ, Shieh JJ, Chung CJ. Association of Sodium-Glucose Transport Protein 2 Inhibitor Use for Type 2 Diabetes and Incidence of Gout in Taiwan. JAMA Netw Open 2021; 4:e2135353
- Fitchett D, Inzucchi SE, Cannon CP, McGuire DK, Scirica BM, Johansen OE, Sambevski S, Kaspers S, Pfarr E, George JT, Zinman B. Empagliflozin Reduced Mortality and Hospitalization for Heart Failure Across the Spectrum of Cardiovascular Risk in the EMPA-REG OUTCOME Trial. Circulation 2019; 139:1384-1395
- Fitchett D, Butler J, van de Borne P, Zinman B, Lachin JM, Wanner C, Woerle HJ, Hantel S, George JT, Johansen OE, Inzucchi SE. Effects of empagliflozin on risk for cardiovascular death and heart failure hospitalization across the spectrum of heart failure risk in the EMPA-REG OUTCOME(R) trial. Eur Heart J 2018; 39:363-370
- Neuen BL, Ohkuma T, Neal B, Matthews DR, de Zeeuw D, Mahaffey KW, Fulcher G, Desai M, Li Q, Deng H, Rosenthal N, Jardine MJ, Bakris G, Perkovic V. Cardiovascular and Renal Outcomes With Canagliflozin According to Baseline Kidney Function. Circulation 2018; 138:1537-1550
- Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu PL, de Zeeuw D, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, Mahaffey KW. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med 2019;
- Jardine MJ, Zhou Z, Mahaffey KW, Oshima M, Agarwal R, Bakris G, Bajaj HS, Bull S, Cannon CP, Charytan DM, de Zeeuw D, Di Tanna GL, Greene T, Heerspink HJL, Levin A, Neal B, Pollock C, Qiu R, Sun T, Wheeler DC, Zhang H, Zinman B, Rosenthal N, Perkovic V. Renal, Cardiovascular, and Safety Outcomes of Canagliflozin by Baseline Kidney Function: A Secondary Analysis of the CREDENCE Randomized Trial. J Am Soc Nephrol2020; 31:1128-1139
- Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Gause-Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med2019; 380:347-357
- Furtado RHM, Bonaca MP, Raz I, Zelniker TA, Mosenzon O, Cahn A, Kuder J, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Nicolau JC, Gause-Nilsson IAM, Fredriksson M, Langkilde AM, Sabatine MS, Wiviott SD. Dapagliflozin and Cardiovascular Outcomes in Patients With Type 2 Diabetes Mellitus and Previous Myocardial Infarction. Circulation 2019; 139:2516-2527
- Kato ET, Silverman MG, Mosenzon O, Zelniker TA, Cahn A, Furtado RHM, Kuder J, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Bonaca MP, Ruff CT, Desai AS, Goto S, Johansson PA, Gause-Nilsson I, Johanson P, Langkilde AM, Raz I, Sabatine MS, Wiviott SD. Effect of Dapagliflozin on Heart Failure and Mortality in Type 2 Diabetes Mellitus. Circulation 2019; 139:2528-2536
- Cahn A, Raz I, Leiter LA, Mosenzon O, Murphy SA, Goodrich EL, Yanuv I, Rozenberg A, Bhatt DL, McGuire DK, Wilding JPH, Gause-Nilsson IAM, Langkilde AM, Sabatine MS, Wiviott SD. Cardiovascular, Renal, and Metabolic Outcomes of Dapagliflozin Versus Placebo in a Primary Cardiovascular Prevention Cohort: Analyses From DECLARE-TIMI 58. Diabetes Care 2021; 44:1159-1167
- Zelniker TA, Bonaca MP, Furtado RHM, Mosenzon O, Kuder JF, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Budaj A, Kiss RG, Padilla F, Gause-Nilsson I, Langkilde AM, Raz I, Sabatine MS, Wiviott SD. Effect of Dapagliflozin on Atrial Fibrillation in Patients With Type 2 Diabetes Mellitus: Insights From the DECLARE-TIMI 58 Trial. Circulation 2020; 141:1227-1234
- Cannon CP, Pratley R, Dagogo-Jack S, Mancuso J, Huyck S, Masiukiewicz U, Charbonnel B, Frederich R, Gallo S, Cosentino F, Shih WJ, Gantz I, Terra SG, Cherney DZI, McGuire DK. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N Engl J Med 2020; 383:1425-1435
- Cosentino F, Cannon CP, Cherney DZI, Masiukiewicz U, Pratley R, Dagogo Jack S, Frederich R, Charbonnel B, Mancuso J, Shih WJ, Terra SG, Cater NB, Gantz I, McGuire DK. Efficacy of Ertugliflozin on Heart Failure-Related Events in Patients with Type 2 Diabetes Mellitus and Established Atherosclerotic Cardiovascular Disease: Results of the VERTIS CV Trial. Circulation 2020;
- McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Belohlavek J, Bohm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukat A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O'Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjostrand M, Langkilde AM. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N Engl J Med2019; 381:1995-2008
- Curtain JP, Docherty KF, Jhund PS, Petrie MC, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Bengtsson O, Langkilde AM, Sjostrand M, Solomon SD, McMurray JJV. Effect of dapagliflozin on ventricular arrhythmias, resuscitated cardiac arrest, or sudden death in DAPA-HF. Eur Heart J 2021;
- Petrie MC, Verma S, Docherty KF, Inzucchi SE, Anand I, Belohlavek J, Bohm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukat A, Ge J, Howlett J, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O'Meara E, Vinh PN, Schou M, Tereshchenko S, Kober L, Kosiborod MN, Langkilde AM, Martinez FA, Ponikowski P, Sabatine MS, Sjostrand M, Solomon SD, Johanson P, Greasley PJ, Boulton D, Bengtsson O, Jhund PS, McMurray JJV. Effect of Dapagliflozin on Worsening Heart Failure and Cardiovascular Death in Patients With Heart Failure With and Without Diabetes. JAMA 2020;
- Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, Januzzi J, Verma S, Tsutsui H, Brueckmann M, Jamal W, Kimura K, Schnee J, Zeller C, Cotton D, Bocchi E, Bohm M, Choi DJ, Chopra V, Chuquiure E, Giannetti N, Janssens S, Zhang J, Gonzalez Juanatey JR, Kaul S, Brunner-La Rocca HP, Merkely B, Nicholls SJ, Perrone S, Pina I, Ponikowski P, Sattar N, Senni M, Seronde MF, Spinar J, Squire I, Taddei S, Wanner C, Zannad F. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N Engl J Med 2020; 383:1413-1424
- Heerspink HJL, Stefansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, Mann JFE, McMurray JJV, Lindberg M, Rossing P, Sjostrom CD, Toto RD, Langkilde AM, Wheeler DC. Dapagliflozin in Patients with Chronic Kidney Disease. N Engl J Med 2020; 383:1436-1446
- Packer M, Butler J, Zannad F, Filippatos G, Ferreira JP, Pocock SJ, Carson P, Anand I, Doehner W, Haass M, Komajda M, Miller A, Pehrson S, Teerlink JR, Schnaidt S, Zeller C, Schnee JM, Anker SD. Effect of Empagliflozin on Worsening Heart Failure Events in Patients With Heart Failure and Preserved Ejection Fraction: EMPEROR-Preserved Trial. Circulation 2021; 144:1284-1294
- Salah HM, Al'Aref SJ, Khan MS, Al-Hawwas M, Vallurupalli S, Mehta JL, Mounsey JP, Greene SJ, McGuire DK, Lopes RD, Fudim M. Effect of sodium-glucose cotransporter 2 inhibitors on cardiovascular and kidney outcomes-Systematic review and meta-analysis of randomized placebo-controlled trials. Am Heart J 2021; 232:10-22
- Voors AA, Angermann CE, Teerlink JR, Collins SP, Kosiborod M, Biegus J, Ferreira JP, Nassif ME, Psotka MA, Tromp J, Borleffs CJW, Ma C, Comin-Colet J, Fu M, Janssens SP, Kiss RG, Mentz RJ, Sakata Y, Schirmer H, Schou M, Schulze PC, Spinarova L, Volterrani M, Wranicz JK, Zeymer U, Zieroth S, Brueckmann M, Blatchford JP, Salsali A, Ponikowski P. The SGLT2 inhibitor empagliflozin in patients hospitalized for acute heart failure: a multinational randomized trial. Nat Med 2022; 28:568-574
- Lytvyn Y, Bjornstad P, Udell JA, Lovshin JA, Cherney DZI. Sodium Glucose Cotransporter-2 Inhibition in Heart Failure: Potential Mechanisms, Clinical Applications, and Summary of Clinical Trials. Circulation 2017; 136:1643-1658
- Inzucchi SE, Zinman B, Fitchett D, Wanner C, Ferrannini E, Schumacher M, Schmoor C, Ohneberg K, Johansen OE, George JT, Hantel S, Bluhmki E, Lachin JM. How Does Empagliflozin Reduce Cardiovascular Mortality? Insights From a Mediation Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2018; 41:356-363
- Ferrannini E. Sodium-Glucose Co-transporters and Their Inhibition: Clinical Physiology. Cell Metab 2017; 26:27-38
- Mudaliar S, Alloju S, Henry RR. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016; 39:1115-1122
- Verma S, McMurray JJV. SGLT2 inhibitors and mechanisms of cardiovascular benefit: a state-of-the-art review. Diabetologia 2018; 61:2108-2117
- Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, Johansen OE, Woerle HJ, Broedl UC, Zinman B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl J Med 2016; 375:323-334
- Cherney DZI, Zinman B, Inzucchi SE, Koitka-Weber A, Mattheus M, von Eynatten M, Wanner C. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: an exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet Diabetes Endocrinol 2017; 5:610-621
- Perkovic V, de Zeeuw D, Mahaffey KW, Fulcher G, Erondu N, Shaw W, Barrett TD, Weidner-Wells M, Deng H, Matthews DR, Neal B. Canagliflozin and renal outcomes in type 2 diabetes: results from the CANVAS Program randomised clinical trials. Lancet Diabetes Endocrinol 2018; 6:691-704
- Neuen BL, Ohkuma T, Neal B, Matthews DR, de Zeeuw D, Mahaffey KW, Fulcher G, Li Q, Jardine M, Oh R, Heerspink HL, Perkovic V. Effect of Canagliflozin on Renal and Cardiovascular Outcomes across Different Levels of Albuminuria: Data from the CANVAS Program. J Am Soc Nephrol 2019; 30:2229-2242
- Mosenzon O, Wiviott SD, Cahn A, Rozenberg A, Yanuv I, Goodrich EL, Murphy SA, Heerspink HJL, Zelniker TA, Dwyer JP, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Kato ET, Gause-Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS, Raz I. Effects of dapagliflozin on development and progression of kidney disease in patients with type 2 diabetes: an analysis from the DECLARE-TIMI 58 randomised trial. Lancet Diabetes Endocrinol 2019;
- Chertow G, Vart P, Jongs N, Toto R, Gorriz JL, Hou FF, McMurray J, Correa-Rotter R, Rossing P, Sjostrom CD, Stefansson B, Langkilde AM, Wheeler D, Heerspink H. Effects of Dapagliflozin in Stage 4 Chronic Kidney Disease. J Am Soc Nephrol 2021;
- Wheeler DC, Stefansson BV, Jongs N, Chertow GM, Greene T, Hou FF, McMurray JJV, Correa-Rotter R, Rossing P, Toto RD, Sjostrom CD, Langkilde AM, Heerspink HJL. Effects of dapagliflozin on major adverse kidney and cardiovascular events in patients with diabetic and non-diabetic chronic kidney disease: a prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol 2021; 9:22-31
- Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Furtado RHM, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Sabatine MS. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019; 393:31-39
- Muskiet MHA, Wheeler DC, Heerspink HJL. New pharmacological strategies for protecting kidney function in type 2 diabetes. Lancet Diabetes Endocrinol 2019; 7:397-412
- Shimizu M, Suzuki K, Kato K, Jojima T, Iijima T, Murohisa T, Iijima M, Takekawa H, Usui I, Hiraishi H, Aso Y. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes Metab 2019; 21:285-292
- Eriksson JW, Lundkvist P, Jansson PA, Johansson L, Kvarnstrom M, Moris L, Miliotis T, Forsberg GB, Riserus U, Lind L, Oscarsson J. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: a double-blind randomised placebo-controlled study. Diabetologia 2018; 61:1923-1934
- Sattar N, Fitchett D, Hantel S, George JT, Zinman B. Empagliflozin is associated with improvements in liver enzymes potentially consistent with reductions in liver fat: results from randomised trials including the EMPA-REG OUTCOME(R) trial. Diabetologia 2018; 61:2155-2163
- Kuchay MS, Krishan S, Mishra SK, Farooqui KJ, Singh MK, Wasir JS, Bansal B, Kaur P, Jevalikar G, Gill HK, Choudhary NS, Mithal A. Effect of Empagliflozin on Liver Fat in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial (E-LIFT Trial). Diabetes Care 2018; 41:1801-1808
- Raj H, Durgia H, Palui R, Kamalanathan S, Selvarajan S, Kar SS, Sahoo J. SGLT-2 inhibitors in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: A systematic review. World J Diabetes 2019; 10:114-132
- Kahl S, Gancheva S, Strassburger K, Herder C, Machann J, Katsuyama H, Kabisch S, Henkel E, Kopf S, Lagerpusch M, Kantartzis K, Kupriyanova Y, Markgraf D, van Gemert T, Knebel B, Wolkersdorfer MF, Kuss O, Hwang JH, Bornstein SR, Kasperk C, Stefan N, Pfeiffer A, Birkenfeld AL, Roden M. Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 2020; 43:298-305
- Akuta N, Watanabe C, Kawamura Y, Arase Y, Saitoh S, Fujiyama S, Sezaki H, Hosaka T, Kobayashi M, Kobayashi M, Suzuki Y, Suzuki F, Ikeda K, Kumada H. Effects of a sodium-glucose cotransporter 2 inhibitor in nonalcoholic fatty liver disease complicated by diabetes mellitus: Preliminary prospective study based on serial liver biopsies. Hepatol Commun 2017; 1:46-52
- Silverii GA, Monami M, Mannucci E. Sodium-glucose co-transporter-2 inhibitors and all-cause mortality: A meta-analysis of randomized controlled trials. Diabetes Obes Metab 2021; 23:1052-1056
- Shi FH, Li H, Yue J, Jiang YH, Gu ZC, Ma J, Lin HW. Clinical Adverse Events of High-Dose vs Low-Dose Sodium-Glucose Cotransporter 2 Inhibitors in Type 2 Diabetes: A Meta-Analysis of 51 Randomized Clinical Trials. J Clin Endocrinol Metab 2020; 105
- Lin DS, Lee JK, Chen WJ. Clinical Adverse Events Associated with Sodium-Glucose Cotransporter 2 Inhibitors: A Meta-Analysis Involving 10 Randomized Clinical Trials and 71 553 Individuals. J Clin Endocrinol Metab 2021; 106:2133-2145
- Puckrin R, Saltiel MP, Reynier P, Azoulay L, Yu OHY, Filion KB. SGLT-2 inhibitors and the risk of infections: a systematic review and meta-analysis of randomized controlled trials. Acta Diabetol 2018; 55:503-514
- Nyirjesy P, Sobel JD, Fung A, Mayer C, Capuano G, Ways K, Usiskin K. Genital mycotic infections with canagliflozin, a sodium glucose co-transporter 2 inhibitor, in patients with type 2 diabetes mellitus: a pooled analysis of clinical studies. Curr Med Res Opin 2014; 30:1109-1119
- Bersoff-Matcha SJ, Chamberlain C, Cao C, Kortepeter C, Chong WH. Fournier Gangrene Associated With Sodium-Glucose Cotransporter-2 Inhibitors: A Review of Spontaneous Postmarketing Cases. Ann Intern Med2019;
- Chia L, Crum-Cianflone NF. Emergence of multi-drug resistant organisms (MDROs) causing Fournier's gangrene. J Infect 2018; 76:38-43
- Dave CV, Schneeweiss S, Patorno E. Association of Sodium-Glucose Cotransporter 2 Inhibitor Treatment With Risk of Hospitalization for Fournier Gangrene Among Men. JAMA Intern Med 2019;
- Nadkarni GN, Ferrandino R, Chang A, Surapaneni A, Chauhan K, Poojary P, Saha A, Ferket B, Grams ME, Coca SG. Acute Kidney Injury in Patients on SGLT2 Inhibitors: A Propensity-Matched Analysis. Diabetes Care2017; 40:1479-1485
- Neuen BL, Young T, Heerspink HJL, Neal B, Perkovic V, Billot L, Mahaffey KW, Charytan DM, Wheeler DC, Arnott C, Bompoint S, Levin A, Jardine MJ. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: a systematic review and meta-analysis. Lancet Diabetes Endocrinol 2019; 7:845-854
- Cahn A, Mosenzon O, Wiviott SD, Rozenberg A, Yanuv I, Goodrich EL, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Gause-Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS, Raz I. Efficacy and Safety of Dapagliflozin in the Elderly: Analysis From the DECLARE-TIMI 58 Study. Diabetes Care2020; 43:468-475
- Fralick M, Schneeweiss S, Patorno E. Risk of Diabetic Ketoacidosis after Initiation of an SGLT2 Inhibitor. N Engl J Med 2017; 376:2300-2302
- Taylor SI, Blau JE, Rother KI. SGLT2 Inhibitors May Predispose to Ketoacidosis. J Clin Endocrinol Metab 2015; 100:2849-2852
- Hamblin PS, Wong R, Ekinci EI, Fourlanos S, Shah S, Jones AR, Hare MJL, Calder GL, Epa DS, George EM, Giri R, Kotowicz MA, Kyi M, Lafontaine N, MacIsaac RJ, Nolan BJ, O'Neal DN, Renouf D, Varadarajan S, Wong J, Xu S, Bach LA. SGLT2 Inhibitors Increase the Risk of Diabetic Ketoacidosis Developing in the Community and During Hospital Admission. J Clin Endocrinol Metab 2019; 104:3077-3087
- Watts NB, Bilezikian JP, Usiskin K, Edwards R, Desai M, Law G, Meininger G. Effects of Canagliflozin on Fracture Risk in Patients With Type 2 Diabetes Mellitus. J Clin Endocrinol Metab 2016; 101:157-166
- Li X, Li T, Cheng Y, Lu Y, Xue M, Xu L, Liu X, Yu X, Sun B, Chen L. Effects of SGLT2 inhibitors on fractures and bone mineral density in type 2 diabetes mellitus: an updated meta-analysis. Diabetes Metab Res Rev2019:e3170
- Bilezikian JP, Watts NB, Usiskin K, Polidori D, Fung A, Sullivan D, Rosenthal N. Evaluation of Bone Mineral Density and Bone Biomarkers in Patients With Type 2 Diabetes Treated With Canagliflozin. J Clin Endocrinol Metab 2016; 101:44-51
- Bolinder J, Ljunggren O, Johansson L, Wilding J, Langkilde AM, Sjostrom CD, Sugg J, Parikh S. Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes Metab 2014; 16:159-169
- Rosenstock J, Frias J, Pall D, Charbonnel B, Pascu R, Saur D, Darekar A, Huyck S, Shi H, Lauring B, Terra SG. Effect of ertugliflozin on glucose control, body weight, blood pressure and bone density in type 2 diabetes mellitus inadequately controlled on metformin monotherapy (VERTIS MET). Diabetes Obes Metab 2018; 20:520-529
- Inzucchi SE, Iliev H, Pfarr E, Zinman B. Empagliflozin and Assessment of Lower-Limb Amputations in the EMPA-REG OUTCOME Trial. Diabetes Care 2018; 41:e4-e5
- Patoulias D, Papadopoulos C, Doumas M. Updated meta-analysis assessing the risk of amputation with sodium-glucose co-transporter-2 inhibitors in the hallmark cardiovascular and renal outcome trials. Diabetes Obes Metab 2021; 23:1063-1065
- Kosiborod MN, Esterline R, Furtado RHM, Oscarsson J, Gasparyan SB, Koch GG, Martinez F, Mukhtar O, Verma S, Chopra V, Buenconsejo J, Langkilde AM, Ambery P, Tang F, Gosch K, Windsor SL, Akin EE, Soares RVP, Moia DDF, Aboudara M, Hoffmann Filho CR, Feitosa ADM, Fonseca A, Garla V, Gordon RA, Javaheri A, Jaeger CP, Leaes PE, Nassif M, Pursley M, Silveira FS, Barroso WKS, Lazcano Soto JR, Nigro Maia L, Berwanger O. Dapagliflozin in patients with cardiometabolic risk factors hospitalised with COVID-19 (DARE-19): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol 2021;
- Rendell MS. Efficacy and safety of sotagliflozin in treating diabetes type 1. Expert Opin Pharmacother 2018; 19:307-315
- Cefalo CMA, Cinti F, Moffa S, Impronta F, Sorice GP, Mezza T, Pontecorvi A, Giaccari A. Sotagliflozin, the first dual SGLT inhibitor: current outlook and perspectives. Cardiovasc Diabetol 2019; 18:20
- Song P, Onishi A, Koepsell H, Vallon V. Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert Opin Ther Targets 2016; 20:1109-1125
- Nuffer W, Williams B, Trujillo JM. A review of sotagliflozin for use in type 1 diabetes. Ther Adv Endocrinol Metab2019; 10:2042018819890527
- Zambrowicz B, Ogbaa I, Frazier K, Banks P, Turnage A, Freiman J, Boehm KA, Ruff D, Powell D, Sands A. Effects of LX4211, a dual sodium-dependent glucose cotransporters 1 and 2 inhibitor, on postprandial glucose, insulin, glucagon-like peptide 1, and peptide tyrosine tyrosine in a dose-timing study in healthy subjects. Clin Ther 2013; 35:1162-1173 e1168
- Powell DR, Zambrowicz B, Morrow L, Beysen C, Hompesch M, Turner S, Hellerstein M, Banks P, Strumph P, Lapuerta P. Sotagliflozin Decreases Postprandial Glucose and Insulin Concentrations by Delaying Intestinal Glucose Absorption. J Clin Endocrinol Metab 2020; 105
- Buse JB, Garg SK, Rosenstock J, Bailey TS, Banks P, Bode BW, Danne T, Kushner JA, Lane WS, Lapuerta P, McGuire DK, Peters AL, Reed J, Sawhney S, Strumph P. Sotagliflozin in Combination With Optimized Insulin Therapy in Adults With Type 1 Diabetes: The North American inTandem1 Study. Diabetes Care 2018; 41:1970-1980
- Danne T, Cariou B, Banks P, Brandle M, Brath H, Franek E, Kushner JA, Lapuerta P, McGuire DK, Peters AL, Sawhney S, Strumph P. HbA1c and Hypoglycemia Reductions at 24 and 52 Weeks With Sotagliflozin in Combination With Insulin in Adults With Type 1 Diabetes: The European inTandem2 Study. Diabetes Care 2018; 41:1981-1990
- Garg SK, Henry RR, Banks P, Buse JB, Davies MJ, Fulcher GR, Pozzilli P, Gesty-Palmer D, Lapuerta P, Simo R, Danne T, McGuire DK, Kushner JA, Peters A, Strumph P. Effects of Sotagliflozin Added to Insulin in Patients with Type 1 Diabetes. N Engl J Med 2017; 377:2337-2348
- Rosenstock J, Cefalu WT, Lapuerta P, Zambrowicz B, Ogbaa I, Banks P, Sands A. Greater dose-ranging effects on A1C levels than on glucosuria with LX4211, a dual inhibitor of SGLT1 and SGLT2, in patients with type 2 diabetes on metformin monotherapy. Diabetes Care 2015; 38:431-438
- Zambrowicz B, Lapuerta P, Strumph P, Banks P, Wilson A, Ogbaa I, Sands A, Powell D. LX4211 therapy reduces postprandial glucose levels in patients with type 2 diabetes mellitus and renal impairment despite low urinary glucose excretion. Clin Ther 2015; 37:71-82 e12
- Bhatt DL, Szarek M, Steg PG, Cannon CP, Leiter LA, McGuire DK, Lewis JB, Riddle MC, Voors AA, Metra M, Lund LH, Komajda M, Testani JM, Wilcox CS, Ponikowski P, Lopes RD, Verma S, Lapuerta P, Pitt B. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N Engl J Med 2021; 384:117-128
- Bhatt DL, Szarek M, Pitt B, Cannon CP, Leiter LA, McGuire DK, Lewis JB, Riddle MC, Inzucchi SE, Kosiborod MN, Cherney DZI, Dwyer JP, Scirica BM, Bailey CJ, Diaz R, Ray KK, Udell JA, Lopes RD, Lapuerta P, Steg PG. Sotagliflozin in Patients with Diabetes and Chronic Kidney Disease. N Engl J Med 2021; 384:129-139
- van Raalte DH, Bjornstad P, Persson F, Powell DR, de Cassia Castro R, Wang PS, Liu M, Heerspink HJL, Cherney D. The Impact of Sotagliflozin on Renal Function, Albuminuria, Blood Pressure, and Hematocrit in Adults With Type 1 Diabetes. Diabetes Care 2019; 42:1921-1929
- Raskin P, Cincotta AH. Bromocriptine-QR therapy for the management of type 2 diabetes mellitus: developmental basis and therapeutic profile summary. Expert Rev Endocrinol Metab 2016; 11:113-148
- Lamos EM, Levitt DL, Munir KM. A review of dopamine agonist therapy in type 2 diabetes and effects on cardio-metabolic parameters. Prim Care Diabetes 2016; 10:60-65
- Holt RI, Barnett AH, Bailey CJ. Bromocriptine: old drug, new formulation and new indication. Diabetes Obes Metab 2010; 12:1048-1057
- Gaziano JM, Cincotta AH, O'Connor CM, Ezrokhi M, Rutty D, Ma ZJ, Scranton RE. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care 2010; 33:1503-1508
- Nauck MA, Meier JJ. The incretin effect in healthy individuals and those with type 2 diabetes: physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol 2016; 4:525-536
- Nauck MA, Meier JJ. Incretin hormones: Their role in health and disease. Diabetes Obes Metab 2018; 20 Suppl 1:5-21
- Brubaker PL, Drucker DJ. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 2004; 145:2653-2659
- Sesti G, Avogaro A, Belcastro S, Bonora BM, Croci M, Daniele G, Dauriz M, Dotta F, Formichi C, Frontoni S, Invitti C, Orsi E, Picconi F, Resi V, Bonora E, Purrello F. Ten years of experience with DPP-4 inhibitors for the treatment of type 2 diabetes mellitus. Acta Diabetol 2019;
- Zhang X, Zhao Q. Effects of dipeptidyl peptidase-4 inhibitors on blood pressure in patients with type 2 diabetes: A systematic review and meta-analysis. J Hypertens 2016; 34:167-175
- Nauck MA, Meier JJ, Cavender MA, Abd El Aziz M, Drucker DJ. Cardiovascular Actions and Clinical Outcomes With Glucagon-Like Peptide-1 Receptor Agonists and Dipeptidyl Peptidase-4 Inhibitors. Circulation 2017; 136:849-870
- Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013; 369:1317-1326
- Scirica BM, Braunwald E, Raz I, Cavender MA, Morrow DA, Jarolim P, Udell JA, Mosenzon O, Im K, Umez-Eronini AA, Pollack PS, Hirshberg B, Frederich R, Lewis BS, McGuire DK, Davidson J, Steg PG, Bhatt DL. Heart failure, saxagliptin, and diabetes mellitus: observations from the SAVOR-TIMI 53 randomized trial. Circulation 2014; 130:1579-1588
- White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Cushman WC, Zannad F. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013; 369:1327-1335
- Zannad F, Cannon CP, Cushman WC, Bakris GL, Menon V, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Lam H, White WB. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 2015; 385:2067-2076
- Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, Josse R, Kaufman KD, Koglin J, Korn S, Lachin JM, McGuire DK, Pencina MJ, Standl E, Stein PP, Suryawanshi S, Van de Werf F, Peterson ED, Holman RR. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2015; 373:232-242
- Rosenstock J, Perkovic V, Johansen OE, Cooper ME, Kahn SE, Marx N, Alexander JH, Pencina M, Toto RD, Wanner C, Zinman B, Woerle HJ, Baanstra D, Pfarr E, Schnaidt S, Meinicke T, George JT, von Eynatten M, McGuire DK. Effect of Linagliptin vs Placebo on Major Cardiovascular Events in Adults With Type 2 Diabetes and High Cardiovascular and Renal Risk: The CARMELINA Randomized Clinical Trial. JAMA 2019; 321:69-79
- McGuire DK, Alexander JH, Johansen OE, Perkovic V, Rosenstock J, Cooper ME, Wanner C, Kahn SE, Toto RD, Zinman B, Baanstra D, Pfarr E, Schnaidt S, Meinicke T, George JT, von Eynatten M, Marx N, Cahn A. Linagliptin Effects on Heart Failure and Related Outcomes in Individuals With Type 2 Diabetes Mellitus at High Cardiovascular and Renal Risk in CARMELINA. Circulation 2019; 139:351-361
- Men P, Li XT, Tang HL, Zhai SD. Efficacy and safety of saxagliptin in patients with type 2 diabetes: A systematic review and meta-analysis. PLoS One 2018; 13:e0197321
- Mosenzon O, Leibowitz G, Bhatt DL, Cahn A, Hirshberg B, Wei C, Im K, Rozenberg A, Yanuv I, Stahre C, Ray KK, Iqbal N, Braunwald E, Scirica BM, Raz I. Effect of Saxagliptin on Renal Outcomes in the SAVOR-TIMI 53 Trial. Diabetes Care 2017; 40:69-76
- Cornel JH, Bakris GL, Stevens SR, Alvarsson M, Bax WA, Chuang LM, Engel SS, Lopes RD, McGuire DK, Riefflin A, Rodbard HW, Sinay I, Tankova T, Wainstein J, Peterson ED, Holman RR. Effect of Sitagliptin on Kidney Function and Respective Cardiovascular Outcomes in Type 2 Diabetes: Outcomes From TECOS. Diabetes Care 2016; 39:2304-2310
- Roshanov PS, Dennis BB. Incretin-based therapies are associated with acute pancreatitis: Meta-analysis of large randomized controlled trials. Diabetes Res Clin Pract 2015; 110:e13-17
- Tkac I, Raz I. Combined Analysis of Three Large Interventional Trials With Gliptins Indicates Increased Incidence of Acute Pancreatitis in Patients With Type 2 Diabetes. Diabetes Care 2017; 40:284-286
- Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG, Tsiotos GG, Vege SS. Classification of acute pancreatitis--2012: revision of the Atlanta classification and definitions by international consensus. Gut2013; 62:102-111
- Saito T, Ohnuma K, Suzuki H, Dang NH, Hatano R, Ninomiya H, Morimoto C. Polyarthropathy in type 2 diabetes patients treated with DPP4 inhibitors. Diabetes Res Clin Pract 2013; 102:e8-e12
- Gentilella R, Pechtner V, Corcos A, Consoli A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: are they all the same? Diabetes Metab Res Rev 2019; 35:e3070
- Frias JP, Bonora E, Nevarez Ruiz L, Li YG, Yu Z, Milicevic Z, Malik R, Bethel MA, Cox DA. Efficacy and Safety of Dulaglutide 3.0 mg and 4.5 mg Versus Dulaglutide 1.5 mg in Metformin-Treated Patients With Type 2 Diabetes in a Randomized Controlled Trial (AWARD-11). Diabetes Care 2021; 44:765-773
- Jones AG, McDonald TJ, Shields BM, Hill AV, Hyde CJ, Knight BA, Hattersley AT. Markers of beta-Cell Failure Predict Poor Glycemic Response to GLP-1 Receptor Agonist Therapy in Type 2 Diabetes. Diabetes Care 2016; 39:250-257
- Wysham CH, Lin J, Kuritzky L. Safety and efficacy of a glucagon-like peptide-1 receptor agonist added to basal insulin therapy versus basal insulin with or without a rapid-acting insulin in patients with type 2 diabetes: results of a meta-analysis. Postgrad Med 2017; 129:436-445
- Abd El Aziz MS, Kahle M, Meier JJ, Nauck MA. A meta-analysis comparing clinical effects of short- or long-acting GLP-1 receptor agonists versus insulin treatment from head-to-head studies in type 2 diabetic patients. Diabetes Obes Metab 2017; 19:216-227
- Castellana M, Cignarelli A, Brescia F, Perrini S, Natalicchio A, Laviola L, Giorgino F. Efficacy and safety of GLP-1 receptor agonists as add-on to SGLT2 inhibitors in type 2 diabetes mellitus: A meta-analysis. Sci Rep 2019; 9:19351
- Tchang BG, Kumar RB, Aronne LJ. Pharmacologic Treatment of Overweight and Obesity in Adults. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Grossman A, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2021.
- Davies MJ, Bergenstal R, Bode B, Kushner RF, Lewin A, Skjoth TV, Andreasen AH, Jensen CB, DeFronzo RA. Efficacy of Liraglutide for Weight Loss Among Patients With Type 2 Diabetes: The SCALE Diabetes Randomized Clinical Trial. JAMA 2015; 314:687-699
- Davies M, Faerch L, Jeppesen OK, Pakseresht A, Pedersen SD, Perreault L, Rosenstock J, Shimomura I, Viljoen A, Wadden TA, Lingvay I. Semaglutide 2.4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet2021; 397:971-984
- Frias JP, Auerbach P, Bajaj HS, Fukushima Y, Lingvay I, Macura S, Sondergaard AL, Tankova TI, Tentolouris N, Buse JB. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol 2021; 9:563-574
- Lorenz M, Lawson F, Owens D, Raccah D, Roy-Duval C, Lehmann A, Perfetti R, Blonde L. Differential effects of glucagon-like peptide-1 receptor agonists on heart rate. Cardiovasc Diabetol 2017; 16:6
- Pfeffer MA, Claggett B, Diaz R, Dickstein K, Gerstein HC, Kober LV, Lawson FC, Ping L, Wei X, Lewis EF, Maggioni AP, McMurray JJ, Probstfield JL, Riddle MC, Solomon SD, Tardif JC. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. N Engl J Med 2015; 373:2247-2257
- Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2016; 375:311-322
- Mann JFE, Fonseca V, Mosenzon O, Raz I, Goldman B, Idorn T, von Scholten BJ, Poulter NR. Effects of Liraglutide Versus Placebo on Cardiovascular Events in Patients With Type 2 Diabetes Mellitus and Chronic Kidney Disease. Circulation 2018; 138:2908-2918
- Verma S, Poulter NR, Bhatt DL, Bain SC, Buse JB, Leiter LA, Nauck MA, Pratley RE, Zinman B, Orsted DD, Monk Fries T, Rasmussen S, Marso SP. Effects of Liraglutide on Cardiovascular Outcomes in Patients With Type 2 Diabetes Mellitus With or Without History of Myocardial Infarction or Stroke. Circulation 2018; 138:2884-2894
- Marso SP, Baeres FMM, Bain SC, Goldman B, Husain M, Nauck MA, Poulter NR, Pratley RE, Thomsen AB, Buse JB. Effects of Liraglutide on Cardiovascular Outcomes in Patients With Diabetes With or Without Heart Failure. J Am Coll Cardiol 2020; 75:1128-1141
- Dhatariya K, Bain SC, Buse JB, Simpson R, Tarnow L, Kaltoft MS, Stellfeld M, Tornoe K, Pratley RE. The Impact of Liraglutide on Diabetes-Related Foot Ulceration and Associated Complications in Patients With Type 2 Diabetes at High Risk for Cardiovascular Events: Results From the LEADER Trial. Diabetes Care 2018; 41:2229-2235
- Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, Woo V, Hansen O, Holst AG, Pettersson J, Vilsboll T. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med 2016; 375:1834-1844
- Holman RR, Bethel MA, Mentz RJ, Thompson VP, Lokhnygina Y, Buse JB, Chan JC, Choi J, Gustavson SM, Iqbal N, Maggioni AP, Marso SP, Ohman P, Pagidipati NJ, Poulter N, Ramachandran A, Zinman B, Hernandez AF. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2017; 377:1228-1239
- Hernandez AF, Green JB, Janmohamed S, D'Agostino RB, Sr., Granger CB, Jones NP, Leiter LA, Rosenberg AE, Sigmon KN, Somerville MC, Thorpe KM, McMurray JJV, Del Prato S. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet 2018; 392:1519-1529
- Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, Probstfield J, Riesmeyer JS, Riddle MC, Ryden L, Xavier D, Atisso CM, Dyal L, Hall S, Rao-Melacini P, Wong G, Avezum A, Basile J, Chung N, Conget I, Cushman WC, Franek E, Hancu N, Hanefeld M, Holt S, Jansky P, Keltai M, Lanas F, Leiter LA, Lopez-Jaramillo P, Cardona Munoz EG, Pirags V, Pogosova N, Raubenheimer PJ, Shaw JE, Sheu WH, Temelkova-Kurktschiev T. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet 2019;
- Gerstein HC, Hart R, Colhoun HM, Diaz R, Lakshmanan M, Botros FT, Probstfield J, Riddle MC, Ryden L, Atisso CM, Dyal L, Hall S, Avezum A, Basile J, Conget I, Cushman WC, Hancu N, Hanefeld M, Jansky P, Keltai M, Lanas F, Leiter LA, Lopez-Jaramillo P, Munoz EGC, Pogosova N, Raubenheimer PJ, Shaw JE, Sheu WH, Temelkova-Kurktschiev T. The effect of dulaglutide on stroke: an exploratory analysis of the REWIND trial. Lancet Diabetes Endocrinol 2020; 8:106-114
- Kristensen SL, Rorth R, Jhund PS, Docherty KF, Sattar N, Preiss D, Kober L, Petrie MC, McMurray JJV. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol 2019; 7:776-785
- Sposito AC, Berwanger O, de Carvalho LSF, Saraiva JFK. GLP-1RAs in type 2 diabetes: mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovasc Diabetol 2018; 17:157
- Lam CSP, Ramasundarahettige C, Branch KRH, Sattar N, Rosenstock J, Pratley R, Del Prato S, Lopes RD, Niemoeller E, Khurmi NS, Baek S, Gerstein HC. Efpeglenatide and Clinical Outcomes With and Without Concomitant Sodium-Glucose Cotransporter-2 Inhibition Use in Type 2 Diabetes: Exploratory Analysis of the AMPLITUDE-O Trial. Circulation 2022; 145:565-574
- Margulies KB, Hernandez AF, Redfield MM, Givertz MM, Oliveira GH, Cole R, Mann DL, Whellan DJ, Kiernan MS, Felker GM, McNulty SE, Anstrom KJ, Shah MR, Braunwald E, Cappola TP. Effects of Liraglutide on Clinical Stability Among Patients With Advanced Heart Failure and Reduced Ejection Fraction: A Randomized Clinical Trial. JAMA 2016; 316:500-508
- Jorsal A, Kistorp C, Holmager P, Tougaard RS, Nielsen R, Hanselmann A, Nilsson B, Moller JE, Hjort J, Rasmussen J, Boesgaard TW, Schou M, Videbaek L, Gustafsson I, Flyvbjerg A, Wiggers H, Tarnow L. Effect of liraglutide, a glucagon-like peptide-1 analogue, on left ventricular function in stable chronic heart failure patients with and without diabetes (LIVE)-a multicentre, double-blind, randomised, placebo-controlled trial. Eur J Heart Fail 2017; 19:69-77
- Fudim M, White J, Pagidipati NJ, Lokhnygina Y, Wainstein J, Murin J, Iqbal N, Ohman P, Lopes RD, Reicher B, Holman RR, Hernandez AF, Mentz RJ. Effect of Once-Weekly Exenatide in Patients With Type 2 Diabetes Mellitus With and Without Heart Failure and Heart Failure-Related Outcomes: Insights From the EXSCEL Trial. Circulation 2019; 140:1613-1622
- Husain M, Bain SC, Jeppesen OK, Lingvay I, Sorrig R, Treppendahl MB, Vilsboll T. Semaglutide (SUSTAIN and PIONEER) reduces cardiovascular events in type 2 diabetes across varying cardiovascular risk. Diabetes Obes Metab 2020; 22:442-451
- Muskiet MHA, Tonneijck L, Huang Y, Liu M, Saremi A, Heerspink HJL, van Raalte DH. Lixisenatide and renal outcomes in patients with type 2 diabetes and acute coronary syndrome: an exploratory analysis of the ELIXA randomised, placebo-controlled trial. Lancet Diabetes Endocrinol 2018; 6:859-869
- Mann JFE, Orsted DD, Brown-Frandsen K, Marso SP, Poulter NR, Rasmussen S, Tornoe K, Zinman B, Buse JB. Liraglutide and Renal Outcomes in Type 2 Diabetes. N Engl J Med 2017; 377:839-848
- Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, Probstfield J, Botros FT, Riddle MC, Ryden L, Xavier D, Atisso CM, Dyal L, Hall S, Rao-Melacini P, Wong G, Avezum A, Basile J, Chung N, Conget I, Cushman WC, Franek E, Hancu N, Hanefeld M, Holt S, Jansky P, Keltai M, Lanas F, Leiter LA, Lopez-Jaramillo P, Cardona Munoz EG, Pirags V, Pogosova N, Raubenheimer PJ, Shaw JE, Sheu WH, Temelkova-Kurktschiev T. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomised, placebo-controlled trial. Lancet 2019;
- Tuttle KR, Lakshmanan MC, Rayner B, Busch RS, Zimmermann AG, Woodward DB, Botros FT. Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): a multicentre, open-label, randomised trial. Lancet Diabetes Endocrinol 2018; 6:605-617
- Shaman AM, Bain SC, Bakris GL, Buse JB, Idorn T, Mahaffey KW, Mann JFE, Nauck MA, Rasmussen S, Rossing P, Wolthers B, Zinman B, Perkovic V. Effect of the Glucagon-Like Peptide-1 Receptor Agonists Semaglutide and Liraglutide on Kidney Outcomes in Patients With Type 2 Diabetes: Pooled Analysis of SUSTAIN 6 and LEADER. Circulation 2022; 145:575-585
- Gluud LL, Knop FK, Vilsboll T. Effects of lixisenatide on elevated liver transaminases: systematic review with individual patient data meta-analysis of randomised controlled trials on patients with type 2 diabetes. BMJ Open2014; 4:e005325
- Armstrong MJ, Houlihan DD, Rowe IA, Clausen WH, Elbrond B, Gough SC, Tomlinson JW, Newsome PN. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: individual patient data meta-analysis of the LEAD program. Aliment Pharmacol Ther 2013; 37:234-242
- Shao N, Kuang HY, Hao M, Gao XY, Lin WJ, Zou W. Benefits of exenatide on obesity and non-alcoholic fatty liver disease with elevated liver enzymes in patients with type 2 diabetes. Diabetes Metab Res Rev 2014; 30:521-529
- Yan J, Yao B, Kuang H, Yang X, Huang Q, Hong T, Li Y, Dou J, Yang W, Qin G, Yuan H, Xiao X, Luo S, Shan Z, Deng H, Tan Y, Xu F, Xu W, Zeng L, Kang Z, Weng J. Liraglutide, Sitagliptin, and Insulin Glargine Added to Metformin: The Effect on Body Weight and Intrahepatic Lipid in Patients With Type 2 Diabetes Mellitus and Nonalcoholic Fatty Liver Disease. Hepatology 2019; 69:2414-2426
- Bouchi R, Nakano Y, Fukuda T, Takeuchi T, Murakami M, Minami I, Izumiyama H, Hashimoto K, Yoshimoto T, Ogawa Y. Reduction of visceral fat by liraglutide is associated with ameliorations of hepatic steatosis, albuminuria, and micro-inflammation in type 2 diabetic patients with insulin treatment: a randomized control trial. Endocr J 2017; 64:269-281
- Kuchay MS, Krishan S, Mishra SK, Choudhary NS, Singh MK, Wasir JS, Kaur P, Gill HK, Bano T, Farooqui KJ, Mithal A. Effect of dulaglutide on liver fat in patients with type 2 diabetes and NAFLD: randomised controlled trial (D-LIFT trial). Diabetologia 2020; 63:2434-2445
- Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, Hazlehurst JM, Guo K, team Lt, Abouda G, Aldersley MA, Stocken D, Gough SC, Tomlinson JW, Brown RM, Hubscher SG, Newsome PN. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016; 387:679-690
- Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, Sanyal AJ, Sejling AS, Harrison SA. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N Engl J Med 2021; 384:1113-1124
- Faillie JL, Yu OH, Yin H, Hillaire-Buys D, Barkun A, Azoulay L. Association of Bile Duct and Gallbladder Diseases With the Use of Incretin-Based Drugs in Patients With Type 2 Diabetes Mellitus. JAMA Intern Med2016; 176:1474-1481
- Monami M, Nreu B, Scatena A, Cresci B, Andreozzi F, Sesti G, Mannucci E. Safety issues with glucagon-like peptide-1 receptor agonists (pancreatitis, pancreatic cancer and cholelithiasis): Data from randomized controlled trials. Diabetes Obes Metab 2017; 19:1233-1241
- He L, Wang J, Ping F, Yang N, Huang J, Li Y, Xu L, Li W, Zhang H. Association of Glucagon-Like Peptide-1 Receptor Agonist Use With Risk of Gallbladder and Biliary Diseases: A Systematic Review and Meta-analysis of Randomized Clinical Trials. JAMA Intern Med 2022; 182:513-519
- Nauck MA, Muus Ghorbani ML, Kreiner E, Saevereid HA, Buse JB, La Macchia O. Effects of Liraglutide Compared With Placebo on Events of Acute Gallbladder or Biliary Disease in Patients With Type 2 Diabetes at High Risk for Cardiovascular Events in the LEADER Randomized Trial. Diabetes Care 2019; 42:1912-1920
- Bethel MA, Patel RA, Merrill P, Lokhnygina Y, Buse JB, Mentz RJ, Pagidipati NJ, Chan JC, Gustavson SM, Iqbal N, Maggioni AP, Ohman P, Poulter NR, Ramachandran A, Zinman B, Hernandez AF, Holman RR. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a meta-analysis. Lancet Diabetes Endocrinol 2018; 6:105-113
- Steinberg WM, Buse JB, Ghorbani MLM, Orsted DD, Nauck MA. Amylase, Lipase, and Acute Pancreatitis in People With Type 2 Diabetes Treated With Liraglutide: Results From the LEADER Randomized Trial. Diabetes Care 2017; 40:966-972
- Liu Y, Tian Q, Yang J, Wang H, Hong T. No pancreatic safety concern following glucagon-like peptide-1 receptor agonist therapies: A pooled analysis of cardiovascular outcome trials. Diabetes Metab Res Rev 2018; 34:e3061
- Singh AK, Gangopadhyay KK, Singh R. Risk of acute pancreatitis with incretin-based therapy: a systematic review and updated meta-analysis of cardiovascular outcomes trials. Expert Rev Clin Pharmacol 2020; 13:461-468
- Vilsboll T, Bain SC, Leiter LA, Lingvay I, Matthews D, Simo R, Helmark IC, Wijayasinghe N, Larsen M. Semaglutide, reduction in glycated haemoglobin and the risk of diabetic retinopathy. Diabetes Obes Metab2018; 20:889-897
- Bethel MA, Diaz R, Castellana N, Bhattacharya I, Gerstein HC, Lakshmanan MC. HbA1c Change and Diabetic Retinopathy During GLP-1 Receptor Agonist Cardiovascular Outcome Trials: A Meta-analysis and Meta-regression. Diabetes Care 2021; 44:290-296
- Avgerinos I, Karagiannis T, Malandris K, Liakos A, Mainou M, Bekiari E, Matthews DR, Tsapas A. Glucagon-like peptide-1 receptor agonists and microvascular outcomes in type 2 diabetes: A systematic review and meta-analysis. Diabetes Obes Metab 2019; 21:188-193
- Hedrington MS, Davis SN. Oral semaglutide for the treatment of type 2 diabetes. Expert Opin Pharmacother2019; 20:133-141
- Avgerinos I, Michailidis T, Liakos A, Karagiannis T, Matthews DR, Tsapas A, Bekiari E. Oral semaglutide for type 2 diabetes: A systematic review and meta-analysis. Diabetes Obes Metab 2019;
- Aroda VR, Rosenstock J, Terauchi Y, Altuntas Y, Lalic NM, Morales Villegas EC, Jeppesen OK, Christiansen E, Hertz CL, Haluzik M. PIONEER 1: Randomized Clinical Trial of the Efficacy and Safety of Oral Semaglutide Monotherapy in Comparison With Placebo in Patients With Type 2 Diabetes. Diabetes Care 2019; 42:1724-1732
- Rosenstock J, Allison D, Birkenfeld AL, Blicher TM, Deenadayalan S, Jacobsen JB, Serusclat P, Violante R, Watada H, Davies M. Effect of Additional Oral Semaglutide vs Sitagliptin on Glycated Hemoglobin in Adults With Type 2 Diabetes Uncontrolled With Metformin Alone or With Sulfonylurea: The PIONEER 3 Randomized Clinical Trial. JAMA 2019; 321:1466-1480
- Pieber TR, Bode B, Mertens A, Cho YM, Christiansen E, Hertz CL, Wallenstein SOR, Buse JB. Efficacy and safety of oral semaglutide with flexible dose adjustment versus sitagliptin in type 2 diabetes (PIONEER 7): a multicentre, open-label, randomised, phase 3a trial. Lancet Diabetes Endocrinol 2019; 7:528-539
- Rodbard HW, Rosenstock J, Canani LH, Deerochanawong C, Gumprecht J, Lindberg SO, Lingvay I, Sondergaard AL, Treppendahl MB, Montanya E. Oral Semaglutide Versus Empagliflozin in Patients With Type 2 Diabetes Uncontrolled on Metformin: The PIONEER 2 Trial. Diabetes Care 2019; 42:2272-2281
- Pratley R, Amod A, Hoff ST, Kadowaki T, Lingvay I, Nauck M, Pedersen KB, Saugstrup T, Meier JJ. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet 2019; 394:39-50
- Davies M, Pieber TR, Hartoft-Nielsen ML, Hansen OKH, Jabbour S, Rosenstock J. Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2017; 318:1460-1470
- Husain M, Birkenfeld AL, Donsmark M, Dungan K, Eliaschewitz FG, Franco DR, Jeppesen OK, Lingvay I, Mosenzon O, Pedersen SD, Tack CJ, Thomsen M, Vilsboll T, Warren ML, Bain SC. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med 2019; 381:841-851
- Bucheit J, Ayers J, Pamulapati L, Browning A, Sisson E. A Novel Dual Incretin Agent, Tirzepatide (LY3298176), for the Treatment of Type 2 Diabetes Mellitus and Cardiometabolic Health. J Cardiovasc Pharmacol 2022; 80:171-179
- Syed YY. Tirzepatide: First Approval. Drugs 2022;
- Nauck MA, Quast DR, Wefers J, Pfeiffer AFH. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: A pathophysiological update. Diabetes Obes Metab 2021; 23 Suppl 3:5-29
- Heise T, Mari A, DeVries JH, Urva S, Li J, Pratt EJ, Coskun T, Thomas MK, Mather KJ, Haupt A, Milicevic Z. Effects of subcutaneous tirzepatide versus placebo or semaglutide on pancreatic islet function and insulin sensitivity in adults with type 2 diabetes: a multicentre, randomised, double-blind, parallel-arm, phase 1 clinical trial. Lancet Diabetes Endocrinol 2022; 10:418-429
- Thomas MK, Nikooienejad A, Bray R, Cui X, Wilson J, Duffin K, Milicevic Z, Haupt A, Robins DA. Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes. J Clin Endocrinol Metab 2021; 106:388-396
- Rosenstock J, Wysham C, Frias JP, Kaneko S, Lee CJ, Fernandez Lando L, Mao H, Cui X, Karanikas CA, Thieu VT. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet 2021; 398:143-155
- Frias JP, Davies MJ, Rosenstock J, Perez Manghi FC, Fernandez Lando L, Bergman BK, Liu B, Cui X, Brown K, SURPASS Investigators. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N Engl J Med 2021; 385:503-515
- Ludvik B, Giorgino F, Jodar E, Frias JP, Fernandez Lando L, Brown K, Bray R, Rodriguez A. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet2021; 398:583-598
- Del Prato S, Kahn SE, Pavo I, Weerakkody GJ, Yang Z, Doupis J, Aizenberg D, Wynne AG, Riesmeyer JS, Heine RJ, Wiese RJ, SURPASS Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet 2021; 398:1811-1824
- Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, Rodriguez A. Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial. JAMA 2022; 327:534-545
- Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, Urva S, Gimeno RE, Milicevic Z, Robins D, Haupt A. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018; 392:2180-2193
- Wilson JM, Nikooienejad A, Robins DA, Roell WC, Riesmeyer JS, Haupt A, Duffin KL, Taskinen MR, Ruotolo G. The dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist, tirzepatide, improves lipoprotein biomarkers associated with insulin resistance and cardiovascular risk in patients with type 2 diabetes. Diabetes Obes Metab 2020; 22:2451-2459
- Feingold KR. Obesity and Dyslipidemia. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2020.
- Sattar N, McGuire DK, Pavo I, Weerakkody GJ, Nishiyama H, Wiese RJ, Zoungas S. Tirzepatide cardiovascular event risk assessment: a pre-specified meta-analysis. Nat Med 2022; 28:591-598
- Gastaldelli A, Cusi K, Fernandez Lando L, Bray R, Brouwers B, Rodriguez A. Effect of tirzepatide versus insulin degludec on liver fat content and abdominal adipose tissue in people with type 2 diabetes (SURPASS-3 MRI): a substudy of the randomised, open-label, parallel-group, phase 3 SURPASS-3 trial. Lancet Diabetes Endocrinol2022; 10:393-406
- Hartman ML, Sanyal AJ, Loomba R, Wilson JM, Nikooienejad A, Bray R, Karanikas CA, Duffin KL, Robins DA, Haupt A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020; 43:1352-1355
- Perreault L, Rodbard H, Valentine V, Johnson E. Optimizing Fixed-Ratio Combination Therapy in Type 2 Diabetes. Adv Ther 2019; 36:265-277
- Billings LK, Doshi A, Gouet D, Oviedo A, Rodbard HW, Tentolouris N, Gron R, Halladin N, Jodar E. Efficacy and Safety of IDegLira Versus Basal-Bolus Insulin Therapy in Patients With Type 2 Diabetes Uncontrolled on Metformin and Basal Insulin: The DUAL VII Randomized Clinical Trial. Diabetes Care 2018; 41:1009-1016
- Cai X, Gao X, Yang W, Ji L. Comparison between insulin degludec/liraglutide treatment and insulin glargine/lixisenatide treatment in type 2 diabetes: a systematic review and meta-analysis. Expert Opin Pharmacother 2017; 18:1789-1798
- Rosenstock J, Aronson R, Grunberger G, Hanefeld M, Piatti P, Serusclat P, Cheng X, Zhou T, Niemoeller E, Souhami E, Davies M. Benefits of LixiLan, a Titratable Fixed-Ratio Combination of Insulin Glargine Plus Lixisenatide, Versus Insulin Glargine and Lixisenatide Monocomponents in Type 2 Diabetes Inadequately Controlled on Oral Agents: The LixiLan-O Randomized Trial. Diabetes Care 2016; 39:2026-2035
- Gough SC, Bode B, Woo V, Rodbard HW, Linjawi S, Poulsen P, Damgaard LH, Buse JB. Efficacy and safety of a fixed-ratio combination of insulin degludec and liraglutide (IDegLira) compared with its components given alone: results of a phase 3, open-label, randomised, 26-week, treat-to-target trial in insulin-naive patients with type 2 diabetes. Lancet Diabetes Endocrinol 2014; 2:885-893
- Younk LM, Davis SN. Evaluation of colesevelam hydrochloride for the treatment of type 2 diabetes. Expert Opin Drug Metab Toxicol 2012; 8:515-525
- Garg A, Grundy SM. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med 1994; 121:416-422
- Prawitt J, Caron S, Staels B. Glucose-lowering effects of intestinal bile acid sequestration through enhancement of splanchnic glucose utilization. Trends Endocrinol Metab 2014; 25:235-244
- Hansen M, Sonne DP, Knop FK. Bile acid sequestrants: glucose-lowering mechanisms and efficacy in type 2 diabetes. Curr Diab Rep 2014; 14:482
- Sonne DP, Hansen M, Knop FK. Bile acid sequestrants in type 2 diabetes: potential effects on GLP1 secretion. Eur J Endocrinol 2014; 171:R47-65
- Beysen C, Murphy EJ, Deines K, Chan M, Tsang E, Glass A, Turner SM, Protasio J, Riiff T, Hellerstein MK. Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: a randomised controlled study. Diabetologia 2012; 55:432-442
- Shapiro H, Kolodziejczyk AA, Halstuch D, Elinav E. Bile acids in glucose metabolism in health and disease. J Exp Med 2018; 215:383-396
- Potthoff MJ, Potts A, He T, Duarte JA, Taussig R, Mangelsdorf DJ, Kliewer SA, Burgess SC. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol 2013; 304:G371-380
- Harach T, Pols TW, Nomura M, Maida A, Watanabe M, Auwerx J, Schoonjans K. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci Rep 2012; 2:430
- Trabelsi MS, Daoudi M, Prawitt J, Ducastel S, Touche V, Sayin SI, Perino A, Brighton CA, Sebti Y, Kluza J, Briand O, Dehondt H, Vallez E, Dorchies E, Baud G, Spinelli V, Hennuyer N, Caron S, Bantubungi K, Caiazzo R, Reimann F, Marchetti P, Lefebvre P, Backhed F, Gribble FM, Schoonjans K, Pattou F, Tailleux A, Staels B, Lestavel S. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat Commun 2015; 6:7629
- Ooi CP, Loke SC. Colesevelam for type 2 diabetes mellitus. Cochrane Database Syst Rev 2012; 12:CD009361
- Feingold KR. Cholesterol Lowering Drugs. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2021.
- The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA 1984; 251:351-364
- The Lipid Research Clinics Coronary Primary Prevention Trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA 1984; 251:365-374
- Younk LM, Mikeladze M, Davis SN. Pramlintide and the treatment of diabetes: a review of the data since its introduction. Expert Opin Pharmacother 2011; 12:1439-1451
- Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta-analysis. Diabetes Obes Metab 2011; 13:169-180
- Riddle M, Pencek R, Charenkavanich S, Lutz K, Wilhelm K, Porter L. Randomized comparison of pramlintide or mealtime insulin added to basal insulin treatment for patients with type 2 diabetes. Diabetes Care 2009; 32:1577-1582
- Chapman I, Parker B, Doran S, Feinle-Bisset C, Wishart J, Strobel S, Wang Y, Burns C, Lush C, Weyer C, Horowitz M. Effect of pramlintide on satiety and food intake in obese subjects and subjects with type 2 diabetes. Diabetologia 2005; 48:838-848