ABSTRACT
Phosphorus is critical to many functions in human biology. Deprivation of phosphorus may manifest as disorders of the musculoskeletal system, reflecting its important role in energy metabolism and skeletal mineralization. Phosphorus excess can promote heterotopic mineralization and is associated with mortality, particularly in the setting of chronic kidney disease. Inorganic phosphorus, primarily occurring as phosphate (PO4), is highly regulated by transport systems in intestine and kidney, and is essential for the formation of a mineralized skeleton. Parathyroid hormone (PTH) and Fibroblast Growth Factor 23 (FGF23) are major hormonal regulators of phosphate homeostasis and membrane abundance of PO4 transporters. Tissue distribution of alpha-klotho permits a primary renal specificity for FGF23 actions. Disorders of phosphate metabolism that are encountered in clinical practice are described in this Endotext chapter, with an emphasis on pathophysiologic processes, diagnostic measures, and treatment. The identification of FGF23 as an important mediator of phosphate homeostasis has brought to light the underlying disease processes in many of these conditions, along with the possibility of novel, physiologic-based therapies.
INTRODUCTION
Phosphorus plays an important role in growth, development, bone formation, acid-base regulation, and cellular metabolism. Inorganic phosphorus exists primarily as the critical structural ion, phosphate (PO4), which serves as a constituent of hydroxyapatite, the mineral basis of the vertebrate skeleton, and at the molecular level, providing the molecular backbone of DNA. Its chemical properties allow its use as a biological energy store as adenosine triphosphate. Additionally, phosphorus influences a variety of enzymatic reactions (e.g., glycolysis) and protein functions (e.g., the oxygen-carrying capacity of hemoglobin by regulation of 2,3-diphosphoglycerate synthesis). Finally, phosphorus is an important signaling moiety, as phosphorylation and dephosphorylation of protein structures serves as an activation signal. Indeed, phosphorus is one of the most abundant components of all tissues, and disturbances in its homeostasis can affect almost any organ system. Most phosphorus within the body is in bone (600-700 g), while the remainder is largely distributed in soft tissue (100-200 g). The plasma contains 11-12 mg/dL of total phosphorus (in both organic and inorganic states) in adults. Inorganic phosphorus (Pi) primarily exists as phosphate (PO4), and is the commonly measured fraction, found in plasma at concentrations averaging 3-4 mg/dl in older children and adults. Plasma Pi concentrations values in children are higher, not infrequently as high as 8 mg/dl in small infants, and gradually declining steeply throughout the first year of life, and further in later childhood to adult values. The organic phosphorus component is primarily found in phospholipids. Although this fraction is not routinely assessed clinically, it comprises approximately two-thirds of the total plasma phosphorus (1). Thus, the term “plasma phosphorus” generally is used when referring to plasma inorganic Pi concentrations, and because plasma inorganic Pi is nearly all in the form of the PO4 ion, the terms phosphorus and phosphate are often interchangeably used in the clinical chemistry laboratory. It should be noted that this terminology can be confusing when using mass units (i.e., mg/dl) as the weight of the phosphorus content of the phosphate is reported, yet “serum phosphate” is often used in the clinic setting. When using molar units the concentration of the phosphate and of the phosphorus are equivalent, and less confusion may arise.
Elaborate mechanisms have evolved to maintain phosphate balance, reflecting the critical role that phosphorus plays in cell and organism physiology. Adaptive changes are manifest by a variety of measurable responses, as modified by metabolic Pi need and exogenous Pi supply. Such regulation maintains the plasma and extracellular fluid phosphorus within a relatively narrow range and depends primarily upon gastrointestinal absorption and renal excretion as adjustable mechanisms to effect homeostasis. Although investigators have recognized a variety of hormones and transporter proteins which influence these various processes, in concert with associated changes in other metabolic pathways, the sensory system, the messenger and the mechanisms underlying discriminant regulation of Pi balance remain incompletely understood.
While long-term changes in Pi balance depend on these variables, short-term changes in Pi concentrations can occur due to redistribution between the extracellular fluid and either bone or cell constituents. Such redistribution results secondary to various mechanisms including: elevated levels of insulin and/or glucose; increased concentrations of circulating catecholamines; respiratory alkalosis; enhanced cell production or anabolism; and rapid bone remineralization.
REGULATION OF PHOSPHORUS HOMEOSTASIS
The majority of ingested phosphorus is absorbed in the small intestine. Active transport is mediated by sodium dependent transporter protein(s), and sodium-independent P-transport also occurs. Hormonal mechanisms regulating Pi homeostasis in the kidney are established in more detail. Indeed, the kidney has long been considered the dominant site of regulation of Pi balance, as renal tubular reclamation of filtered Pi occurs in response to complex regulatory mechanisms. Although the fate of Pi has generally been considered a matter of renal elimination, incorporation into organic forms in proliferating cells, or deposition into the mineral phase of bone as hydroxyapatite, the role of intestinal phosphate transport warrants further study. Indeed, it appears that presentation of Pi to the intestine can affect systemic phosphate handling before changes in serum Pi concentration are evident. Moreover, in the setting of severe phosphorus deprivation, the phosphate contained in bone mineral provides a source of phosphorus for the metabolic needs of the organism. The specific roles that the intestine and kidney play in this complex process are discussed below.
Gastrointestinal Absorption Of Phosphorus
Studies of Pi absorption in the intestine have yielded variable results, in part due to confounding influences of nutritional status, the effects of anesthesia on gut transit, species differences, and potential effects of studying whole organisms as opposed to isolated bowel segments. The small intestine is the dominant site of Pi absorption; in mice Pi is absorbed along the entire length of small bowel, but at the highest rate in the ileum. In rats, duodenum and jejunum provide the primary sites of Pi absorption, whereas very little occurs in ileum. This is felt to be more consistent with the pattern of Pi absorption in humans, however studies are subject to the confounding issues noted above. In normal adults net Pi absorption is a linear function of dietary Pi intake. For a dietary Pi range of 4 to 30 mg/kg/day, the net Pi absorption averages 60 to 65% of the intake (2). Intestinal Pi absorption occurs via two routes (Figure 1), a cellularly mediated sodium-dependent active transport mechanism, and sodium independent transport, which is not well characterized. Mechanisms for the latter have been attributed in part to paracellular pathways (3), however recent findings suggest that for glucose, this pathway only accounts for 1-2% of passive glucose uptake in the intestine, far less that once speculated. Finally, the relative roles of these processes appear to be age dependent (4). Animal models suggest that weanling mice have both greater intestinal P transport (and greater expression of NaPi-IIb) than at other ages, presumably to support skeletal growth.
Controversy exists as to what proportion of intestinal Pi absorption is absorbed via sodium-dependent mechanisms and what proportion is sodium-independent. In this regard, the major Na+-dependent phosphate cotransporter identified in intestinal brush border membranes is NPT2b, a member of the SLC34 solute carrier family, also referred to as type II sodium-phosphate cotransporters (5). Earlier studies suggested that approximately 50% of intestinal P transport occurs by sodium-dependent mechanisms, and that most of this activity can be accounted for by the sodium-dependent transporter NPT2b. A lesser contribution to sodium-independent transport has been attributed to either type III transporters (PiT1 and PiT2, see below), and other unknown mechanisms. NPT2b is also expressed in lung, colon, testis/epididymis, liver, and in mammary and salivary glands, with most abundant expression in mammary glands (4). NPT2b is electrogenic, maintains a 3:1 stoichiometry of Na: Pi, prefers the divalent P species, and has a high affinity for Pi binding (6-10). Depending upon species and bowel segment, NPT2b transporters can be regulated by 1,25 dihydroxyvitamin D (), FGF23 (¯), low Pi diet (), and acute phosphate loading (). Energy for the electrochemical uphill process is provided by the sodium gradient, which is maintained by sodium-potassium ATPase. The phosphate incorporated into intestinal cells by this mechanism is ferried from the apical pole to the basolateral pole likely through restricted channels such as the microtubules. Exit of Pi from the enterocyte across the basolateral membrane and into the circulation is a poorly understood process. More recently widely-expressed members of the SLC20 solute carrier family, the type III sodium-phosphate cotransporters PiT1 and PiT2, have been found to be variably expressed in the intestine (11), and PiT2 primarily in ileum. Pit1 and Pit2 prefer to transport the monovalent Pi species (HPO4 -), and maintain a 2 Na: 1 P stoichiometry. These transporters may play a greater role in adaptive responses to intestinal Pi transport than previously recognized. PiT1 upregulates in response to phosphate deprivation, but in a relatively slow time frame, whereas expression of PiT2 and NPT2b upregulate within 24 hrs (12). In NPT2b-/- mice there is approximately 10% sodium-dependent Pi transport activity, suggesting that the type III transporters are of limited significance in the intestine in murine models. The process is further complicated by significant effects of alkaline pH as inhibitory to intestinal Pi transport, Moreover, the adaptation to P deprivation occurs with greater rates of transport occurring at more acidic pH (12) Given the variable nature and segment-specific regulation of NPT2b, the ultimate impact on overall phosphate homeostasis appears to be less well understood at the intestine than at the kidney. The presence of different classes of transporters in the intestine provide for Pi transport under a variety of different conditions such as variable pH, species of PO4 substrate, and Pi supply. Indeed, recent work leads to speculation that much of the adaptive response to intestinal phosphate transport likely occurs by yet unrecognized transporters or transport processes (12).
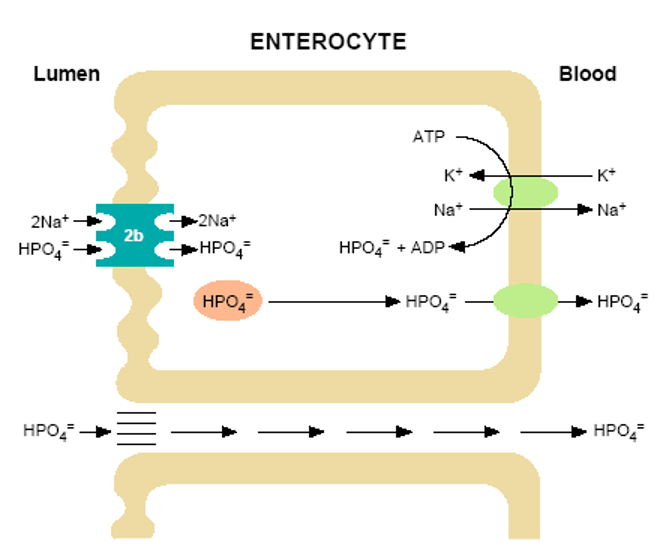
Figure 1. Model of inorganic phosphate (HPO4=) transport in the intestine. At the luminal surface of the enterocyte the brush border membrane harbors sodium-dependent phosphate transporters of the NPT2b type. NPT2b transporters are electrogenic, have high affinity for Pi, and a stoichiometry of 3 Na ions: 1 phosphate. Energy for this transport process is provided by an inward downhill sodium gradient, maintained by transport of Na+ from the cell via a Na+/K+ ATPase cotransporter at the basolateral membrane. The HPO4= incorporated into the enterocytes by this mechanism is transferred to the circulation by poorly understood mechanisms. Type III sodium-dependent transporters are also expressed on the intestinal luminal surface (PiT1 and PiT2) and contribute to this process. Considerable HPO4= absorption occurs via a sodium-independent process(es) such diffusional absorption across the intercellular spaces in the intestine. Other processes have also been hypothesized.
As most diets contain an abundance of Pi, the quantity absorbed nearly always exceeds the need. Factors which may adversely influence the non-regulable, sodium-independent process are the formation of nonabsorbable calcium, aluminum or magnesium phosphate salts in the intestine and age, which reduces Pi absorption by as much as 50%.
Renal Excretion Of Phosphorus
The kidney responds rapidly to changes in serum Pi levels or to dietary Pi intake. The balance between the rates of glomerular filtration and tubular reabsorption (13) determines net renal handling of Pi. Pi concentration in the glomerular ultrafiltrate is approximately 90% of that in plasma, as not all of the plasma Pi is ultrafilterable (14). Since the product of the serum Pi concentration and the glomerular filtration rate (GFR) approximates the filtered load of Pi, a change in the GFR may influence Pi homeostasis if uncompensated by commensurate changes in tubular reabsorption.
The major site of phosphate reabsorption is the proximal convoluted tubule, at which 60% to 70% of reabsorption occurs (Figure 2). Along the proximal convoluted tubule, the transport is heterogeneous, with greatest activity in the S1 segment. Further, increasing, but not conclusive, data supports the existence of a Pi reabsorptive mechanism in the distal tubule. Currently, however, conclusive proof for tubular secretion of Pi in humans is lacking (15).
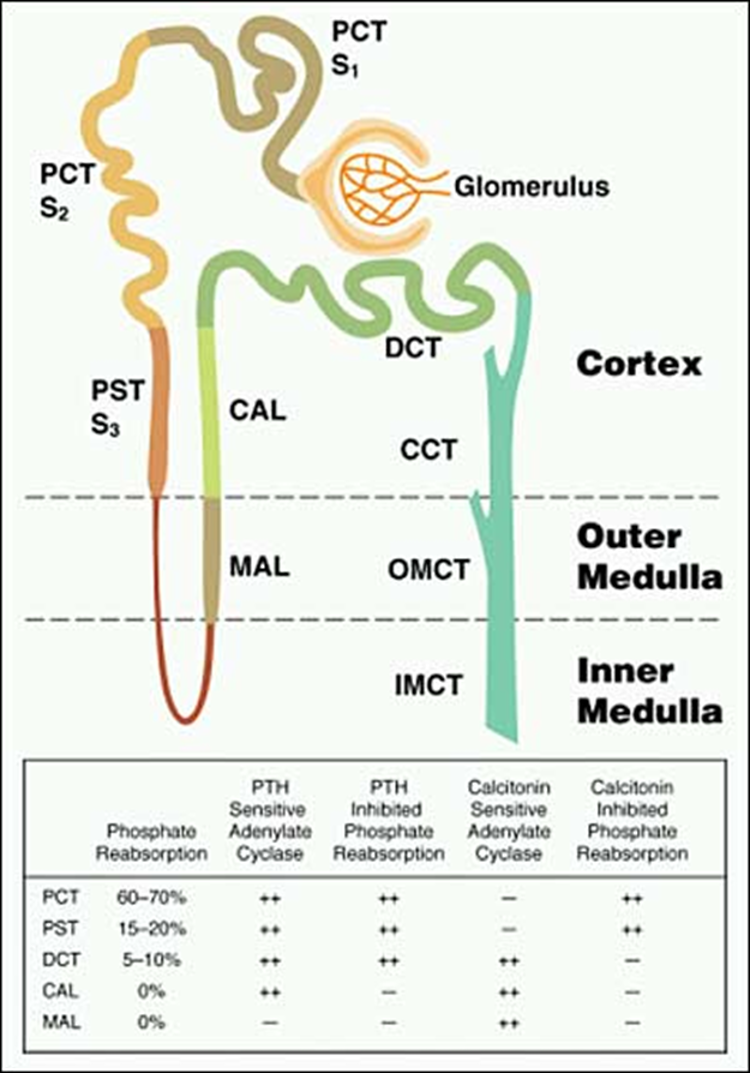
Figure 2. Distribution of Pi reabsorption and hormone-dependent adenylate cyclase activity throughout the renal tubule. The renal tubules consist of a proximal convoluted tubule (PCT), composed of an S1, S2 and S3 segment, a proximal straight tubule (PST), also known as the S3 segment, the loop of Henle, the medullary ascending limb (MAL), the cortical ascending limb (CAL), the distal convoluted tubule (DCT) and three segments of the collecting tubule: the cortical collecting tubule (CCT); the outer medullary collecting tubule (OMCT); and the inner medullary collecting tubule (IMCT). Pi reabsorption occurs primarily in the PCT but is present is the PST and DCT, sites at which parathyroid hormone (PTH) dependent adenylate cyclase is localized. In contrast, calcitonin alters Pi transport at sites devoid of calcitonin dependent adenylate cyclase, suggesting that Pi reabsorption in response to this stimulus occurs by a distinctly different mechanism.
At all three sites of Pi reabsorption, the proximal convoluted tubule, proximal straight tubule and distal tubule, PTH has been shown to decrease Pi reabsorption either by a cAMP-dependent process, or in some cases a cAMP-independent signaling mechanism. In contrast, calcitonin-sensitive adenylate cyclase maps to the medullary and cortical thick ascending limbs and the distal tubule (Figure 2) (16). Although calcitonin has been shown to inhibit Pi reabsorption in proximal convoluted and straight tubules by a cAMP-independent mechanism, the physiologic importance of this action is likely limited. It appears that the major regulators of renal tubular phosphate retention are PTH and the endocrine fibroblast growth factor, FGF23 (see below).
MECHANISM OF PHOSPHATE TRANSPORT
Investigations of the cellular events involved in Pi movement from the renal tubule luminal fluid to the peritubular capillary blood indicate that Pi reabsorption occurs principally by a unidirectional process that proceeds transcellularly. Entry of Pi into the tubular cell across the luminal membrane proceeds by way of a saturable active transport system that is sodium-dependent (analogous to the sodium-dependent co-transport in the intestine) (Figure 3). The rate of Pi transport is dependent on the abundance of transporters functioning in the membrane, and the magnitude of the Na+gradient maintained across the luminal membrane. This gradient depends on the Na+/ATPase or sodium pump on the basolateral membrane. The rate limiting step in transcellular transport is likely the Na+-dependent entry of Pi across the luminal membrane, a process with a low Km for luminal phosphate (~0.43M) which permits highly efficient transport.
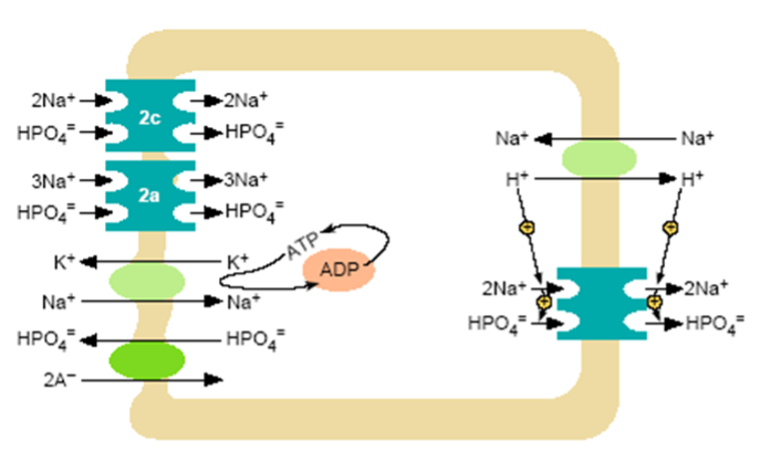
Figure 3. Model of inorganic phosphate transcellular transport in the proximal tubule. At the brush border a Na+/H+ exchanger and NPT2 co-transporters operate. Nearly all proximal tubular reabsorption can be accounted for by the SLC34 (type II) family of sodium-dependent Pi transporters. In mice, NPT2a appears to be the more abundant transporter; it is electrogenic with a 3:1 (Na: PO4) stoichiometry, preferentially transporting the divalent phosphate anion. The lesser abundant NPT2c transporter is electroneutral with a 2:1 (Na: PO4) stoichiometry, but also prefers the divalent phosphate species. In humans NP2c appears to have a more significant role than in mice. The HPO4- that enters the cell across the luminal surface mixes with the intracellular pool of Pi and is transported across the basolateral membrane. This process is poorly understood, but anion exchange mechanisms have been suggested. A Na+/K+ ATPase located on the basolateral membrane pumps Na+ out of the cell maintaining the inward downhill Na gradient, which serves as the driving force for luminal entry of Na+.
The phosphate that enters the tubule cell plays a major role in governing various aspects of cell metabolism and function and is in rapid exchange with intracellular phosphate. Under these conditions the relatively stable free Pi concentration in the cytosol implies that Pi entry into the cell across the brush border membrane must be tightly coupled with either subcellular compartmentalization, organification, or exit across the basolateral membrane (Figure 3). The transport of phosphate across the basolateral membrane is poorly understood, however, several P transport pathways have been postulated, including Na+-Pi cotransport via type III Na-Pi cotransporters, passive diffusion, and anion exchange. The XPR1 transporter has been implicated in transport of phosphate out of cells but the significance of its role in total body P homeostasis is uncertain in humans (17). One animal study provides reasonable evidence for a critical role for this transporter in generalized tubular function (18). In any case, the basolateral Pi transport serves at least two functions: 1) complete transcellular Pi reabsorption when luminal Pi entry exceeds the cellular Pi requirements; and 2) basolateral Pi influx if apical Pi entry is insufficient to satisfy cellular requirements (19).
Pi entry into renal epithelium is primarily performed by the type II class of Na-Pi cotransporters (SLC34 family members), although recently the finding of type III transporters (SLC20 family members, PiT1 and PiT2) in kidney have raised the possibility of a potential role for this class as well (20).These two families of Na-Pi cotransporters share no significant homology in their primary amino acid sequence and as noted above, exhibit substantial variability in substrate affinity, pH dependence and tissue expression. The NPT2 class of transporters account for the bulk of regulated phosphate transport in kidney, and disruption of this regulation may result in significant disease, documenting their physiological importance (21, 22). As with intestinal Pi transport, physiologic differences between these families of Pi transporters provides for functional diversity allowing the body to transfer Pi between compartments in a variety of situations. Of the class II transporters NPT2a and NPT2c transporters are the predominant actors in the proximal renal tubule. NPT2a, the more abundant species in mice, is electrogenic with a 3:1 (Na: PO4) stoichiometry, preferentially transporting the divalent phosphate anion, and has a high affinity for Pi (all features of the NPT2b member of this family, the predominant intestinal sodium-dependent Pi transporter, see above). NPT2c differs from its type 2a/b family members in that is electroneutral with a 2:1 (Na: PO4) stoichiometry, but also prefers the divalent phosphate species. It has a much lower affinity for Pi, but is an efficient transporter due to its electroneutrality. An aspartic acid residue (Asp 224 in human NaPi-IIa) in a sodium binding site within a conserved amino acid cluster in the electrogenic transporters NPT2a and NPT2b, appears to be critical for electrogenicity. It is replaced with a glycine residue (Gly 196 in human NPT2c) in the electroneutral type IIc transporter (23).
Initial attention focused on NPT2a, as it was determined to be the most abundant Na-Pi cotransporter in kidney. Molecular and/or genetic suppression of NPT2a supports its role in mediating brush-border membrane Na-Pi cotransport. Intravenous injection of specific antisense oligonucleotides reduces brush-border membrane Na-Pi cotransport activity in accord with a decrease in NPT2a protein (24). In addition, disruption of the gene encoding NPT2a in mice (Slc34a1) leads to a 70% reduction in brush-border Na-Pi cotransport rate and complete loss of the protein (25, 26). However, the NPT2c transporter may have a relatively more important role for Pi transport in humans as compared to rodents, and appears to have a more widespread tissue distribution. The identification of a unique form of hypophosphatemia, Hereditary Hypophosphatemic Rickets with Hypercalciuria (HHRH) as a loss-of-function mutation in NPT2c has demonstrated an important physiologic role in humans for this transporter (27).
The roles of type III transporters in this process are not established at this time, and the previously described class of type I sodium-dependent phosphate transporters (of the SLC17 family) are not specific Pi transporters and do not appear to be central to the regulation of phosphate homeostasis.
REGULATION OF RENAL TUBULAR PHOSPHATE HANDLING
Several hormones and metabolic perturbations are able to modulate phosphate reabsorption by the kidney. Among these FGF23, PTH, PTHrP, calcitonin, atrial natriuretic peptide, acidosis, TGFb, glucocorticoids, hypercalcemia, and phosphate loading inhibit renal phosphate reclamation (for review, see reference 28). In contrast, IGF-1, growth hormone, insulin, thyroid hormone, EGF, alkalosis, hypocalcemia, and phosphate deprivation (depletion) stimulate renal phosphate reabsorption. The central role of FGF23 in this regard, revealed by the study of clinical disorders of renal phosphate wasting. Indeed, PTH and FGF23 are likely the two most important regulators of renal tubular phosphate handling, and are discussed in greater detail below. The common target for regulation by these factors is the renal proximal tubular cell. Effects of 1,25(OH)2D are less clearly delineated, and such effects in vivo may be mediated by PTH or FGF23.
PTH
Investigations of classical PTH effects on proximal tubule phosphate transport indicate that both the cAMP-protein kinase A (PKA) and the phospholipase C-protein kinase C (PKC) signal transduction pathways are able to modulate this process. The PTH mediated inhibition of phosphate reabsorption operates through the PKC system at low hormone concentrations (10-8 to 10-10 M) and via PKA at higher concentrations. The PKA pathway is the more important mediator of PTH’s role on P handling at the kidney. PTH, after interaction with its receptor, PTHR1, effects a rapid and irreversible endocytosis of NaPi-IIa transporters to the lysosomal compartment, where subsequent proteolytic degradation occurs (29). Stabilization of NPT2a is mediated by NHERF1 which is phosphorylated by PTH’s activation of the PKA and PKC pathways. NHERF3 also binds to NPT2a, but it does not appear to be necessary for apical retention of the transporter.
In contrast to NPT2a, NPT2c transporters are not targeted to lysosomes and their removal from the apical membrane may not be entirely irreversible (30, 31). Although recovery of NPT2a cotransport activity following PTH inhibition requires protein synthesis, this may not be the case for NPT2c. In addition, the abundance of NPT2a-specific mRNA is not changed by parathyroidectomy but is minimally decreased in response to PTH administration. These data implicate PTH as a regulator of renal Na-Pi cotransport in an acute time frame, and that the regulation is determined by changes in the abundance of NaPi-II proteins in the renal brush border membrane (32). Certain aspects of Pi homeostasis at the renal level, however, are not explained by actions of PTH. For instance, even in the setting where parathyroid glands have been removed, regulation of renal P transport by dietary P content still exists, implying that other mediators of this process are at work.
FGF23
FGF23 is the most recently identified important physiologic regulator of renal Pi excretion (33). This novel member of the fibroblast growth factor (FGF) family is produced by osteocytes and osteoblasts, thereby serving as a mechanism by which skeletal mineral demands can be communicated to the kidney, and influencing phosphate economy of the entire organism. In rodents and humans, after days of dietary phosphate loading, circulating FGF23 levels increase, and similarly, with dietary Pi deprivation, FGF23 levels decrease (34). FGF23 activates FGF receptors on the basolateral membrane of renal tubules resulting in removal of type II sodium-dependent Pi transporters from the apical surface of the tubular cell by a NHERF1 dependent process, similar to the mechanism described for PTH above. However, in contrast to PTH, FGF23 actions are mediated activation of ERK1/2 rather than the PTH driven PKA dependent pathway. Evidence also exists for decreased expression of type II sodium-dependent Pi via genomic mechanisms. FGF23 interacts with its receptor via a mechanism now identified as characteristic of the endocrine FGFs. FGF23 recognizes its cognate FGFR only in the presence of the co-receptor, alpha-klotho (35). Activation of this complex results in downstream ERK phosphorylation, and subsequently reduced expression of NaPi-IIa and NaPi-IIc, and CYP27B1 (1-hydroxylase), with an increase in expression of CYP24A1 (24-hydroxylase). This mechanism of signaling is apparent for the endocrine FGFs, FGF19 and FGF21, which require a separate member of the klotho family (beta-klotho) for specific tissue specific activation of FGFRs (for detailed review, see reference 36).
FGF23 contains a unique C-terminal domain, thought to be the site of the interaction with klotho. The FGF-like domain, N-terminal to a furin protease recognition site, is the basis for the interaction of FGF23 with FGFR. Alpha-klotho appears to be able to associate with “c” isoforms of FGFR1 and FGFR3, and also FGFR4 (35). Renal signaling is thought to occur via FGFR1c, thereby rendering the reduced expression of the apical membrane NaPi-II transporters. FGF23 also may play a role movement of transporters from the apical membrane; PTH may play a modulatory or necessary role for this effect (37). The physiologic importance of this system has been demonstrated in several ways. First, mice overexpressing FGF23 demonstrate increased renal Pi clearance and concomitant hypophosphatemia (38). Secondly, FGF23 null mice retain P at the kidney and are hyperphosphatemic (39). Thirdly, administration to mice of an FGF23 neutralizing antibody increases serum Pi (40).
Nevertheless, gaps in our understanding of this pathway remain. Alpha-klotho appears to be more abundantly expressed in distal renal tubules as compared to proximal tubular sites. Thus, the mechanism by which this pathway effects the transporters in the proximal tubule is unclear. Most recently klotho alone has been shown to increase expression of FGF23, and appears to be able to reduce renal tubular phosphate reabsorption, independent of FGF23 (41). These findings are consistent with a unique case of hypophosphatemia associated with a mutation in the klotho region resulting in overexpression of the protein and an abundance of circulating klotho (42). Finally, recent evidence indicating that it certain tissues (heart) FGF signaling may occur in the absence of alpha-klotho, although the physiologic significance of this finding is not certain (43).
The actions of FGF23 and other related proteins as mediators of disease are discussed in detail in the section on Pathophysiology of XLH (see below). Other potential regulators of renal Pi handling have been suggested. These include fragments of matrix extracellular glycoprotein (MEPE), secreted frizzled related protein-4 (sFRP4), stanniocalcin, and other FGFs, including FGF2, and FGF7 (44-47).
The Osteocyte As A Coordinating Center For Phosphate Homeostasis
Osteocytes are distributed throughout lamellar bone in an organized array with interconnections occurring through small tunneling caniculi (for review, see reference 48). Cellular processes extending from the cell body of the osteocyte pass through these caniculi and serve as a means of communication with other cells and to bony surfaces. Interestingly, many of the proteins involved in phosphate regulation are secreted by the osteocyte, including: 1) PHEX, which regulates FGF23 secretion, with loss-of-function resulting in elevated circulating FGF23; 2) DMP1, a SIBLING protein, in which loss-of-function also results in elevated circulating FGF23; 3) FGF23 itself, and 4) FGFR1 which appears to be activated in osteocytes resulting in elevated FGF23 expression. These observations have led to the consideration that the osteocyte may directly respond to phosphate nutritional status, and the osteocytic network throughout the skeleton may relay the mineral demands for bone maintenance to the kidney, where phosphate conservation is regulated. The osteocyte’s response to phosphate status does not appear to be an acute process, as that observed with the extracellular calcium sensing receptor system that regulates PTH secretion in PT glands. The coordination of certain specific matrix proteins may play a role in the local regulation of phosphate supply and mineralization. For instance, skeletal pyrophosphate (PPi) has been identified in increased abundance in the perilacunar bone of Hyp mice, suggesting a potential role of this potent inhibitor of mineralization in the skeletal pathophysiology of the disease (49). Others have demonstrated aberrations in osteopontin in skeletal matrix (50). It follows that genetic disruption of this pathway may result in the profound systemic disturbances observed in the diseases discussed herein.
In sum, repeated observations have confirmed that the balance between urinary excretion and dietary input of Pi is maintained in normal humans, in patients with hyper- and hypoparathyroidism, and under man conditions. This is predominantly due to the ability of the renal tubule to adjust Pi reabsorption rate according to the body’s Pi supply and demand. Thus, Pi reabsorption is increased under conditions of greater need, such as rapid growth, pregnancy, lactation and dietary restriction. Conversely, in times of surfeit, such as slow growth, chronic renal failure or dietary excess, renal Pi reabsorption is curtailed. Such changes in response to chronic changes in Pi availability are characterized by parallel changes in Na-phosphate cotransporter activity, the NPT2 mRNA level and NPT2 protein abundance. These changes are likely mediated by FGF23, as well as other possible factors. Removal of NPT2 cotransporters from the apical membrane of renal tubular cells is an acute process, mediated by PTH. The interaction of these two agents on the overall process may also be important. Indeed, ablation of PTH in a murine model of excess FGF23 abrogates hypophosphatemia. Likewise, suppression of PTH may reduce phosphate losses even with persistence of high FGF23 (51, 52), suggesting an interaction between the two pathways at the renal tubule (53).
Primary disorders of phosphate homeostasis are listed in Table 1. Phosphate abnormalities may also occur in the setting of chronic kidney disease, as effects of therapeutic agents, and nutritional or intestinal absorption problems. Not surprisingly, since the kidney is the primary regulatory site for phosphate homeostasis, aberrant phosphate metabolism results most commonly from altered renal Pi handling. Moreover, the majority of the primary diseases are phosphate-losing disorders in which renal Pi wasting and hypophosphatemia predominate and osteomalacia and rickets are characteristic. Osteomalacia and rickets are disorders of calcification characterized by defects of bone mineralization in adults and bone and cartilage mineralization during growth. In osteomalacia, there is a failure to normally mineralize the newly formed organic matrix (osteoid) of bone. In rickets, a disease of children, there is not only abnormal mineralization of bone but defective cartilage growth plate calcification at the epiphyses as well. Apoptosis of chondrocytes in the hypertrophic zone is reduced, typically resulting in an expanded hypertrophic zone, delayed mineralization and vascularization of the calcification front, with an overall appearance of a widened and disorganized growth plate (54).
The remainder of this chapter reviews the pathophysiology of hypophosphatemic rachitic and osteomalacic disorders, and provides a systematic approach to the diagnosis and management of these diseases. The discussion will focus on disorders in which primary disturbances in phosphate homeostasis occur, emphasizing X-linked hypophosphatemic rickets/osteomalacia (XLH). Other FGF23-mediated disorders including autosomal dominant and autosomal recessive hypophosphatemic rickets (ADHR, ADHR, ARHR1, ARHR2, ARHR3), and tumor-induced osteomalacia (TIO) will be discussed. Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) and Dent's disease will be described as examples of FGF23-independent hypophosphatemia.
|
Table 1. Primary Disorders of Phosphate Homeostasis
|
|
|
Gene
|
Mutation
|
|
FGF23-mediated Hypophosphatemia (XLH)
Autosomal dominant hypophosphatemic rickets (ADHR)
Autosomal recessive hypophosphatemic rickets 1 (ARHR1)
Autosomal recessive hypophosphatemic rickets 2 (ARHR2)
Autosomal recessive hypophosphatemic rickets 2/Raine
syndrome related hypophosphatemia (ARHR3)
McCune-Albright syndrome/fibrous dysplasia
Osteoglophonic dysplasia
Jansen metaphyseal chondrodysplasia
Klotho overexpression
Epidermal nevus syndrome (ENS)/Cutaneous Skeletal
Hypophosphatemia Syndrome (CSHS)
Opsismodysplasia
Tumor-induced osteomalacia (TIO)
|
PHEX
FGF23
DMP1
ENPP1
FAM20C
GNAS1
FGFR1
PTH1R
9;13 translocation
HRAS, NRAS
INPPL1
|
LOF*
GOF*
LOF
LOF
LOF
GOF (somatic)
GOF
GOF
GOF
GOF (somatic)
LOF
|
|
FGF23-Independent
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) (NaPi-IIc deficiency)
Dent’s disease (X-linked recessive hypophosphatemic)
NPT2a deficient Fanconi syndrome
Fanconi-Bickel syndrome.
Hypophosphatemia with osteoporosis and nephrolithiasis
|
SLC34A3
CLCNS
SLC34A1
SLC2A2
SLC34A1/SLC9A3R1
|
LOF
LOF
LOF
LOF
LOF
|
|
Hyperphosphatemia
Hyperphosphatemic tumoral calcinosis
|
GALNT3, FGF23, KLOTHO
|
LOF
|
Mineralization Of Bone And Cartilage
Mineralization of bone is a complex process in which a calcium-phosphate mineral phase is deposited in a highly ordered fashion within the organic matrix (55). Apart from the availability of calcium and phosphorus, requirements for normal mineralization include: 1) adequate metabolic and transport function of chondrocytes and osteoblasts to regulate the concentration of calcium, phosphorus and other ions at the calcification sites; 2) the presence of collagen with unique type, number and distribution of cross-links, distinct patterns of hydroxylation and glycosylation and abundant phosphate content, which collectively facilitate deposition of mineral at gaps (or "hole zones") between the distal ends of collagen molecules; 3) a low concentration of mineralization inhibitors (such as pyrophosphates and proteoglycans) in bone matrix; and 4) maintenance of an appropriate pH of approximately 7.6 for deposition of calcium-phosphate complexes.
The abnormal mineralization in the hypophosphatemic disorders, is due most likely to the limited availability of phosphorus at calcification sites and, in some cases, paracrine inhibitory factors, which result in accumulation of unmineralized osteoid, a sine qua non for the diagnosis of osteomalacia. Since the resultant abundant osteoid is not unique to osteomalacia, establishing the diagnosis of osteomalacia requires dynamic histopathologic demonstration that abnormal mineralization, and not increased production, underlies the observed excess accumulation of osteoid (56, 57). Static histomorphometrical parameters seen in osteomalacia include an increase in osteoid volume and thickness, an increase in bone forming surface covered by incompletely mineralized osteoid, and a decrease in the mineralization front (the percentage of osteoid-covered bone-forming surface undergoing calcification). The critical dynamic parameter used to confirm that osteoid accumulation is due to osteomalacia is the mineral apposition rate.
Inadequate growth plate cartilage mineralization in rickets is primarily observed in the hypertrophic zone of chondrocytes. Irregular alignment and more extensive disorganization of the growth plate may be evident with increasing severity of disease. Calcification in the interstitial regions of this hypertrophic zone is defective. Grossly, these changes result in increased thickness of the epiphyseal plate, and an increase in transverse diameter that often extends beyond the ends of the bone and causes characteristic cupping or flaring.
X-LINKED HYPOPHOSPHATEMIC RICKETS/OSTEOMALACIA
X-linked hypophosphatemic rickets/osteomalacia (XLH) was initially recognized in the 1930s as a form of “vitamin D resistant" and only later, as disorder of renal phosphate wasting. The disorder is inherited in X-linked dominant fashion and is manifest biochemically by a low renal threshold maximum for renal tubular phosphate reabsorption, consequent hypophosphatemia, and low, or inappropriate circulating levels of 1,25(OH)2D. Known biochemical characteristics of XLH and other hypophosphatemic disorders are shown in Table 2. Characteristic features of the disease include growth retardation, osteomalacia and rickets in growing children. The clinical expression of the disease is widely variable, ranging from mild skeletal abnormalities to severe bone disease. Most would agree that a wide spectrum of phenotypic severity occurs in both males (with a mutated gene on their only X chromosome) and females (who are heterozygous for the defective X-linked gene), although clinical experience suggests that females, particularly with certain mutations may express less severe disease (58). Bowing of the lower extremities is usually the first physical sign of the disorder, but is not often evident until 1-2 yrs of age, after the child is standing or walking (59). Biochemical evidence of disease can be detected shortly after birth, however may not become apparent until several weeks to months of age. Short stature generally becomes evident after the first year of life, as well (60), coincident with the timing of bow deformities. Growth abnormalities and limb deformities are both more evident in the lower extremities, since they represent the fastest growing body segment before puberty.
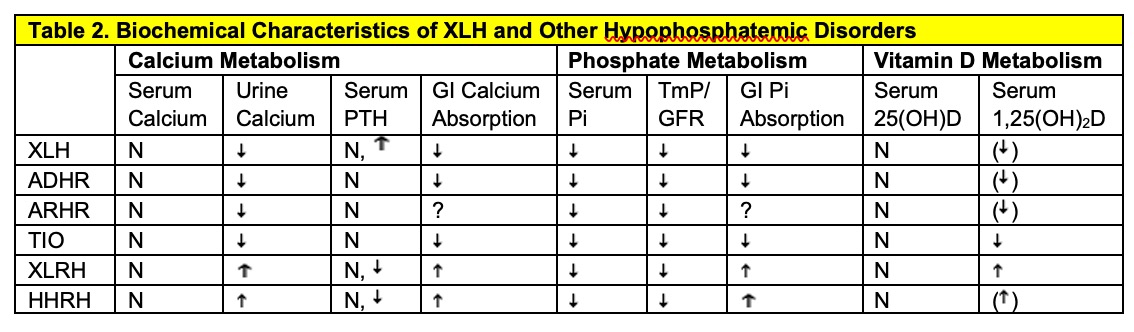
XLH, X-linked hypophosphatemia; ADHR, Autosomal dominant hypophosphatemic rickets; ARHR, Autosomal recessive hypophosphatemic rickets; TIO, Tumor-induced osteomalacia; XLHR, X-linked recessive hypophosphatemia (Dent's Disease); HHRH, Hereditary hypophosphatemic rickets with hypercalciuria. N, normal; , decreased; , increased, ( ), decreased relative to the serum phosphorus concentration; ?, unknown.
The majority of affected children exhibit clinical evidence of rickets (Figure 4), varying from enlargement of the wrists and/or knees to severe malalignment defects such as bowing or knock-knee deformities. (Figure 4). Such defects may result in waddling gait and leg length abnormalities (61). X-ray examination reveals expanded areas of non-mineralized cartilage in epiphyseal regions and lateral curvature of the femora and/or tibia. Strikingly absent are features observed in vitamin D deficiency rickets attributable to hypocalcemia, such as, tetany and convulsions. Muscle weakness and pain are not usually presentations of XLH in early childhood, but emerge later in life.
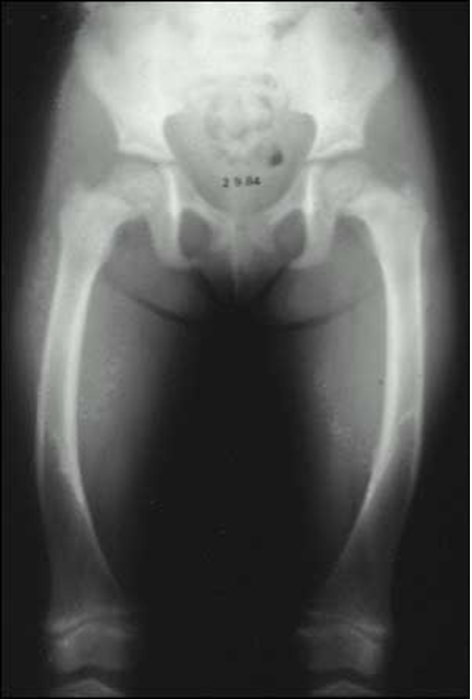
Figure 4. Radiograph of the lower extremities in a patient with X-linked hypophosphatemia. Bowing of the femurs is evident bilaterally. The distal femoral metaphysis is cupped, frayed and widened, radiographic features of an expanded and disorganized growth plate.
Additional signs of the disease may include delayed dentition and dental abscesses (62, 63), which are usually manifest clinically by pain and a gingival papule at the site of involvement. Radiographically there is an enlarged air compartment seen around the root of the affected tooth and an enlarged pulp chamber. Other dental findings that may play a role in the process include impaired mineralization of the dentine compartment of the tooth, and diminished cementum. Craniofacial structural anomalies may also result in crowding of teeth, requiring orthodontic management. Indeed, suture fusion of the cranial bones is aberrant, and craniosynostosis to some degree occurs frequently, and in severe cases require neurosurgical intervention.
Adults with XLH manifest a broad spectrum of disease. They may be asymptomatic or present with severe bone pain. On clinical examination they often display evidence of post-rachitic deformities, such as bowed legs or short stature. However, overt biochemical changes such as elevated serum alkaline phosphatase activity or other biomarkers of bone turnover may not be evident. Adult patients frequently demonstrate features of "active" osteomalacia, characterized radiographically by pseudofractures, coarsened trabeculation, rarified areas and/or non-union fractures, and although variably present, may have elevated serum alkaline phosphatase activity. Symptoms at presentation may reflect the end-result of chronic changes, and may not correlate with apparent current activity of the disease. In spite of marked variability in the clinical presentation of the disease, bone biopsy in affected children and adults nearly always reveals osteomalacia without osteopenia (Figure 5). Histomorphometry of biopsy samples usually demonstrates a reduced rate of formation, diffuse patchy hypomineralization, a decrease in mineralizing surfaces and characteristic areas of hypomineralization of the periosteocytic lacunae (64). Of note, as noted above, increased skeletal PPi identified in the perilacunar bone of Hyp mice, the syngeneic animal model of XLH, may serve to inhibit mineralization locally as well (49).
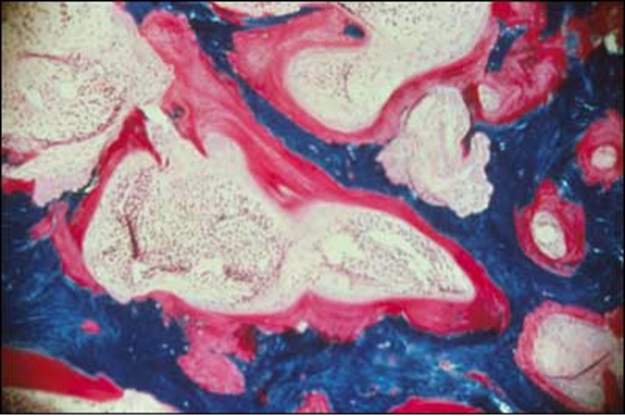
Figure 5. Section from an undecalcified bone biopsy in an untreated patient with X-linked hypophosphatemia. The Goldner stain reveals mineralized bone (blue/green) and an abundance of unmineralized osteoid (red) covering a substantial portion of the surfaces. The width of the osteoid seams is substantially increased.
Osteophytes and other findings of a mineralizing enthesopathy (65) occur frequently and may result in the most severe clinical symptomatology in adulthood. A great deal of the morbidity of XLH in adults arises from the high incidence of arthritis, calcified entheses, and osteophytes. Enthesopathy generally is first detectable radiographically by late in the second decade, or early in the third decade. Older subjects have more sites of involvement, and generally increasing involvement with age; the frequency of involvement appears to be greater in males. With progressive enthesopathy and bony overgrowth, excruciating pain may occur, particularly with fusion of the sacroiliac joint(s) and spinal stenosis (66). These manifestations do not appear to be affected for the better or worse with respect to exposure to currently available therapies (67). It is peculiar that XLH represents a deficiency of mineralization at many skeletal sites, and pathologic ectopic mineralization elsewhere. This paradoxical situation raises the possibility that aberrant humoral factors, in addition to the ambient hypophosphatemia, may play a role in the discordant mineralization abnormalities observed.
Clinical Biochemistry
As previously noted, the primary biochemical abnormality of XLH is hypophosphatemia due to increased urinary phosphate excretion. Moreover, mild gastrointestinal phosphate malabsorption is present in the majority of patients, which may contribute to the evolution of the hypophosphatemia (Table 2) (68, 69).
In contrast, the serum calcium concentration in affected subjects is normal despite gastrointestinal malabsorption of calcium. However, as a consequence of this defect, urinary calcium is often decreased. The severe secondary hyperparathyroidism that occurs in vitamin D deficiency is not present as the degree of calcium malabsorption is not a severe in that condition. However, mildly elevated circulating levels of PTH occur in many patients naive to therapy, and thought to represent the inadequate production of 1,25(OH)2D. Other non-specific but typical findings include elevated serum alkaline phosphatase activity. Serum alkaline phosphatase activity, although usually elevated to 2-3 times the upper limit of normal in childhood, is generally less than the levels observed in nutritional rickets. As noted above, circulating PTH levels may be normal to modestly elevated in naïve patients, but treatment with phosphate salts often aggravate this tendency such that persistent secondary hyperparathyroidism may occur. Because of variability in adulthood, this measure is not a reliable marker of disease involvement in the older age group. Prior to the initiation of therapy, serum 25-OHD levels are normal, and serum 1,25(OH)2D levels are in the low normal range (70, 71). The paradoxical occurrence of hypophosphatemia and normal serum calcitriol levels in affected subjects is consistent with aberrant regulation of both synthesis and clearance of this metabolite (due to increased 25-OHD-24-hydroxylase activity) (72, 73). Circulating levels of FGF23 are generally elevated in individuals with XLH, although overlap may occur. Thus, caution should be applied when using this measure as a strict diagnostic criterion for the diagnosis of XLH, as some subjects have been shown to have circulating FGF23 levels within the normal range, and commercially available assays (which recognize “intact” species or both intact and C-terminal species) do not always provide concordant results.
Genetics
With the recognition that hypophosphatemia is the definitive marker for XLH, Winters et al (74) and Burnett et al (75) discovered that this disease is transmitted as an X-linked dominant disorder. Analysis of data from 13 multigenerational pedigrees identified PHEX (for phosphate regulating gene with homologies to endopeptidases located on the X chromosome) as the gene disrupted in XLH (76). PHEX is located on chromosome Xp22.1, and encodes a 749-amino acid protein with three putative domains: 1) a small amino-terminal intracellular tail; 2) a single, short transmembrane domain; and 3) a large carboxyterminal extracellular domain, containing ten conserved cysteine residues and a HEXXH pentapeptide motif, which characterizes many zinc metalloproteases. Further studies have revealed that PHEX is homologous to the M13 family of membrane-bound metalloproteases, or neutral endopeptidases. M13 family members, including neutral endopeptidase 24.11 (NEP), endothelin-converting enzymes 1 and 2 (ECE-1 and ECE-2), the Kell blood group antigen (KELL), neprilysin-like peptide (NL1), and endothelin converting enzyme-like 1 (ECEL1), degrade or activate a variety of peptide hormones. In addition, like other neutral endopeptidases, immunofluorescent studies have revealed a cell-surface location for PHEX in an orientation consistent with a type II integral membrane glycoprotein (77). It has been demonstrated that certain missense variants in PHEX that substitute a highly conserved cysteine residue will interfere with normal trafficking of the molecule to the plasma membrane (78). Thus, it appears that one mechanism associated with the pathophysiology of XLH is to prevent PHEX from locating to the cell membrane. Other missense variants have been shown in in vitro studies to disrupt enzymatic function of the protein, alter its conformation, or disrupt cellular processing in other ways (79, 80).
Studies in rodents have demonstrated that Phex is predominantly expressed in bones (in osteoblasts/osteocytes) and teeth (in odontoblasts/ameloblasts) (81-84); mRNA, protein or both have also been found in lung, brain, muscle, gonads, skin and parathyroid glands. Subcellular locations appear to be the plasma membrane, endoplasmic reticulum and Golgi organelle. Immunohistochemistry studies suggest that Phex is most abundant on the cell surface of the osteocyte. In sum, the ontogeny of Phex expression suggests a possible role in mineralization in vivo.
Recently combined efforts of many investigators and genetic sequencing laboratories have documented over 850 pathogenic PHEX variants (85). Most of these (>70%) are predicted to generate a truncated PHEX protein. Overall, frameshift, splicing, copy-number, nonsense, and missense variants have been described, and are predicted to cause loss of function of the PHEX protein. A recently updated on-line database of PHEX variants can be accessed at: https://www.rarediseasegenes.com/
The location of Phex expression in bone cells have led to the hypothesis that diminished PHEX/Phex expression in bone initiates the cascade of events responsible for the pathogenesis of XLH. In order to confirm this possibility, several investigators have used targeted over-expression of Phex in attempts to normalize osteoblast mineralization, in vitro, and rescue the Hyp phenotype in vivo (86-88). Results from these studies have not resulted in a complete skeletal rescue, raising questions as to the role of early developmental expression of PHEX, or at least the success of expression when targeted with osteocalcin or type I collagen promoters. Nevertheless, partial rescue of the mineralization defect in Hyp mice occurs, suggesting that local effects of the PHEX mutation may play some role in the mineralization process, but cannot completely restore the skeleton to normality. Of note, this partial rescue occurs in concert with a reduction in FGF23 levels, although not lowered to a truly normal range (88).
In sum, although a physiologic substrate for PHEX has not been identified, the consequence of loss-of-function of PHEX is an elevation in the circulating FGF23 level. Failure of targeted osteoblastic PHEX overexpression to completely rescue Hyp mice may reflect that critical sites (or developmental timing) for PHEX expression are not effectively generated with these models to effectively rescue the skeletal phenotype; this effect may be dependent upon the resultant capacity in these transgenic models of normal PHEX to reduce FGF23 production in mutant cells. ASARM peptides, fragments of SIBLING (small integrin binding ligand N-glycated) proteins, have been shown to inhibit mineralization and potentially play a role in modulation of renal P transport; these peptides have also been shown to be degraded by PHEX (89). Other evidence has suggested that expression of osteopontin expression may be altered in the context of PHEX loss of function as well, (50).
Pathophysiology
Hypophosphatemia in XLH results from the impaired renal proximal tubule function of Pi reabsorption. For some time, XLH was thought to be a primary disorder of the renal tubule, however the consideration that humoral mediation of phosphate wasting in XLH was suggested by two novel clinical findings. First, the persistence of renal phosphate wasting after renal transplantation in a man with XLH indicated a new donor kidney continued to manifest the defect (90). Second, the clinical course of a similar phosphate-wasting syndrome, Tumor Induced Osteomalacia (also referred to as Oncogenic Osteomalacia), resolved upon removal of a tumor, suggesting that the tumor was the source of a mediating factor. Further evidence for humoral mediation was provided by classical parabiosis experiments, suggested that a cross-circulating factor could mediate renal phosphate wasting (91), and by renal cross-transplantation between Hyp and normal mice. These experiments demonstrated continued normal renal phosphate handling after transplantation of Hyp kidney to a normal host, as well as the failure to correct the mutant phenotype upon introduction of a normal kidney to a Hyp host (92). These findings, most consistent with humoral mediation of the Pi wasting in the disease, led to the search for candidate mediators of renal phosphate handling, and eventually to the discovery that FGF23 is an important regulator of renal phosphate homeostasis. Subsequently mean circulating FGF23 concentrations were found to be greater in XLH patients than in unaffected control subjects, providing evidence for the role of this endocrine FGF in XLH.
Renal tubular wasting occurs on the basis of a decreased abundance of NPT2 transporters in the proximal convoluted tubule cells (93-95), and in turn, this reduction in NPT2 abundance is mediated by increased circulating levels of FGF23 (see above, Regulation of Renal Tubular Phosphate Handling). Increased FGF23 occurs in the context of disruption of PHEX, which, like FGF23, is primarily expressed in osteocytes, and FGF23 appears to be produced in a phosphate-sensitive manner.
It remains unclear as to how the loss-of-function of PHEX results in elevated FGF23 levels. The hypothesis that PHEX (a member of the M13 family of zinc-dependent type II cell surface membrane metalloproteinases) could serve as a processor of a phosphaturic hormone such as FGF23 has not been borne out, and the role PHEX plays in this pathway is not clear. Several other phosphate wasting disorders have been described (see below) in which elevated FGF23 occurs in the setting of (presumably) normal PHEX. Such conditions include TIO, where overproduction of FGF23 results in a comparable Pi wasting phenotype. In Autosomal Dominant Hypophosphatemic Rickets (ADHR) specific mutations in FGF23 result in gain of function of the protein (96, 97). The specific mutations disrupt an RXXR protease recognition site, and thereby protect FGF23 from proteolysis, resulting in reduced clearance and elevating circulating levels of this protein, with coincident renal Pi wasting. In yet another genetic disorder, Autosomal Recessive Hypophosphatemic Rickets type I (98), due to homozygous loss of function mutations in dentin matrix protein-1 (DMP1), renal tubular Pi wasting occurs in the setting of increased FGF23 levels. DMP1 is a matrix protein of the SIBLING (small integrin binding ligand N-glycated) family, and, like PHEX and FGF23, has been primarily identified in osteocytes.
In Autosomal Recessive Hypophosphatemic Rickets type II, due to mutations in ENPP1, elevated FGF23 concentrations occur (99, 100). ENPP1 encodes a phosphatase with a critical local role in mineralization, serving to generate the mineralization inhibitor, pyrophosphate (PPi); loss of function of ENPP1 may result in Generalized Arterial Calcification of Infancy (GACI), a fatal disease of infants in which rampant vascular mineralization occurs (101). These findings have suggested the hypothesis that loss of the mineralization inhibitor PPi prompts a signal to compensate for the severe excess vascular mineralization, and increasing FGF23 levels results in an attempt to induce renal phosphate excretion and to limit further excessive mineralization. Nearly all patients who have survived GACI develop renal phosphate wasting and often consequent rickets (102).
Furthermore, FGF23 levels are elevated in mice with biallelic disruptions of DMP1 and with biallelic loss of ENPP1. Transgenic mice which overexpress FGF23, exhibit retarded growth, hypophosphatemia, decreased (or inappropriately normal) serum 1,25(OH)2D levels and rickets/osteomalacia, all features of XLH. Indeed, murine models of all of these disorders (XLH, ADHR, TIO, and ARHR) similarly demonstrate elevated circulating FGF23 levels with concomitant renal phosphate wasting
In sum, enhanced FGF23 activity is common to several phosphate-wasting disorders. In particular, those disorders that share the combined defects of inappropriately low circulating levels of 1,25(OH)2D and renal tubular Pi wasting are associated with increased FGF23 levels. This coincidence of findings holds for XLH, ADHR, ARHR (types I, II, and III), and TIO, and are consistent with the notion that FGF23 is a both a direct regulator of Pi homeostasis at the renal level, a down-regulator of 1a-hydroxylase activity, responsible for the catalysis of 25-OH vitamin D to its active form, and stimulus for its clearance via the 24-hydroxylation pathway. The teleological appeal to this argument stems from the provision of 2 major Pi regulating hormones in the body: firstly, PTH (primarily responsive to serum Ca levels), which also serves to increase Ca levels via an increase in circulating 1,25(OH)2D, and secondly, FGF23 (primarily responsive to Pi), which counters PTH’s calcemic effect by reducing 1,25(OH)2D levels (Figure 6).
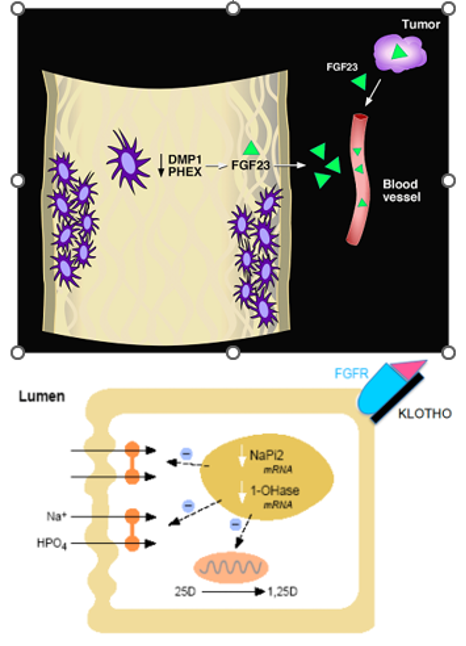
Figure 6. Scheme for the speculated pathophysiology of XLH, ARHR, TIO, and ADHR. Upper panel, osteocytes, comprising a network of connected cells embedded in mineralized bone are the cellular source of PHEX (which is mutated in XLH), DMP1 (which is mutated in ARHR), and FGF23 (which is found in high concentrations in all four of these hypophosphatemic disorders). It follows that loss of PHEX or DMP1 results in increased FGF23 production/secretion by mechanisms that are not currently understood. Circulating FGF23 concentrations may also occur secondary to the increased production associated with various tumors. Lower panel, circulating FGF23 interacts with an FGF receptor (presumably FGFR1) on the basolateral surface of the proximal renal tubular cell. Klotho, produced by the distal renal tubule in both membrane bound and secretory forms, is necessary for the FGF23/FGFR interaction. Signaling through this pathway results in a decrease in NPT2 mRNAs, thereby reducing the abundance of Pi cotransporters on the apical membrane as well as a more rapid action of translocating the transporter off of the apical membrane. Thus, impairment of renal tubular Pi reabsorption results. Likewise, synthesis of 1,25(OH)2D is impaired, while its clearance is augmented. In XLH and ARHR, increased production of FGF23 occurs in the skeleton; in TIO, increased production of FGF23 occurs in tumors; in ADHR, enhanced activity of FGF23 occurs as a result of the specific mutations that retard its metabolic clearance.
Other recent findings have provided support for the role of klotho in the FGF23-mediated hypophosphatemia pathway. An unusual patient with renal tubular Pi wasting and abnormally increased serum klotho has been described (42). Investigation revealed a translocation breakpoint disrupting the region upstream of that encoding klotho. Indeed, mice with disruption of the klotho gene manifest hyperphosphatemia and elevated circulating 1,25(OH)2D levels (103). The proof that klotho is distal to PHEX in this regulatory pathway was shown by crossing the klotho disrupted mice with Hyp (PHEX-deletion) mice. The double mutant (Hyp/Kl-/-) mice were hyperphosphatemic, with elevated 1,25(OH)2D levels, despite having extremely elevated circulating FGF23 levels due to PHEX loss-of-function (104). The unexpected finding that overexpression of klotho can upregulate FGF23 production has also been reported (105).
Indeed, further evidence for the central role of FGF23 in the Pi-regulating process comes from the investigation of another group of rare disorders of Pi homeostasis in which renal Pi conservation is excessive in the setting of increased circulating Pi levels. This group of disorders, known as hyperphosphatemic tumoral calcinosis (HTC), is manifest clinically by precipitation of amorphous calcium-phosphate crystals in soft tissues. This phenomenon is thought to result from an increase in the ambient Ca x Pi solubility product, and occurs as a direct result of enhanced renal tubular reabsorption of Pi (106). In addition, circulating 1,25(OH)2D levels are inappropriately in the high-normal to high range. Thus, the precise converse of primary metabolic derangements occurs, as compared to the XLH-related group of diseases. Initially, HTC was been shown to directly result from loss of function mutations in GALNT3, a glycosylating enzyme important for appropriate O-glycosylation of proteins. This modification appears to be necessary for efficient Golgi secretion of full length FGF23 (107). Interestingly patients with HTC due to GALNT3 mutations have increased circulating levels of the inactive C-terminal fragment of FGF23, but low circulating levels of intact active form of FGF23 (108). Recent evidence implicates that variant post-translational modification of FGF23 can also be modulated by FAM20C: mutations in this gene can result in elevated FGF23 levels, renal phosphate wasting and hypophosphatemia, and referred to as Autosomal Recessive Hypophosphatemic Rickets, type III (ARHR3) and may have clinical features described as Raine syndrome (see table 1) (109, 110).
HTC may also occur in the setting of loss-of-function mutations of FGF23 (111). As with GALNT3-related HTC, these patients have low intact FGF23 level. Loss of function of klotho has also been described in a case of HTC, despite the finding of elevated FGF23 levels, thus rendering the FGF23 inactive at the renal proximal tubule (112). As with hypophosphatemia syndromes, animal models have confirmed the physiologic implications of these clinical scenarios: FGF23 null mice develop a hyperphosphatemic, calcifying phenotype with elevated 1,25(OH)2D levels (39), similar to mice with disruption of the klotho gene (103, 113). As noted above, the klotho protein is now known to be an essential co-factor in FGFR1c activation when FGF23 serves as the activating ligand (35).
The overall physiologic importance of this regulating system requires further study. It is not clear how PHEX or DMP1 result in elevated FGF23 levels. The intriguing aspect of the osteocyte as a potential central cell in this pathway also bears further study.
Treatment
Decades ago, physicians employed pharmacological doses of vitamin D as the cornerstone for treatment of XLH. However, long-term observations indicate that this therapy fails to cure the disease and poses the serious problem of recurrent vitamin D intoxication and renal damage. Indeed, such treatment results only in incomplete healing of the rachitic abnormality, while hypophosphatemia and impaired growth remain. Similar unresponsiveness is typical with use of 25(OH)D.
With the recognition that phosphate depletion is an important contributor to impaired skeletal mineralization, physicians began to devise treatment strategies that employed oral phosphate supplementation to compensate for the renal phosphate wasting and thereby increasing the available Pi to the mineralizing skeleton. This strategy was somewhat successful in terms of improving skeletal lesions, although it was soon realized that pharmacologic amounts of vitamin D were necessary in combination with phosphate supplements to counter the exacerbation of hyperparathyroidism observed in this setting. Such combination therapy was found to be more effective than either administering vitamin D or phosphate alone. With the recognition that circulating 1,25(OH)2D levels are not appropriately regulated in XLH, the use of this metabolite in combination with phosphate was subsequently used to treat the disease (67, 114-116). The current treatment strategy directly addresses the combined calcitriol and phosphorus deficiency characteristic of the disorder. Although this combination therapy has become the conventional therapy for XLH, complete healing of the skeletal lesions is usually not the case, and late complications of the disease are persistent and often debilitating.
In children the goal of therapy is to improve growth velocity, normalize any lower extremity defects, and heal the attendant bone disease. Generally, the treatment regimen includes a period of titration to achieve a maximum dose of 1,25(OH)2D3 (Rocaltrol® or calcitriol), 20-50 ng/kg/day in two divided doses, and phosphorus, (20-50 mg/kg/day, to a maximum of 1-2 gms/day) in 3-5 divided doses.
Use of 1,25(OH)2D3/phosphorus combination therapy involves a significant risk of toxicity. Hypercalcemia, hypercalciuria, renal calcinosis, and hyperparathyroidism can be sequelae of unmonitored therapy. Detrimental effects on renal function were particularly common prior to the frequent monitoring now generally employed with this therapy. Indeed, hypercalcemia, severe nephrocalcinosis and/or diminished creatinine clearance necessitates appropriate dose adjustment, and in some cases discontinuation of therapy. Throughout the treatment course careful attention to renal function, as well as serum and urine calcium is extremely important. Nevertheless, the improved outcome of this therapeutic intervention compared to that achieved by previous regimens, justifies its use, albeit requires an aggressive clinical monitoring schedule.
While such combined therapy often improves growth velocity, refractoriness to the growth-promoting effects of treatment can be encountered in children who present with markedly short stature prior to 4 years of age. For that reason the use of recombinant growth hormone as additional treatment has been suggested (117), however this approach has not been universally recommended in view of the lack of definitive benefits in controlled studies, and a risk of resultant worsening of the disproportional stature (118), although others have not identified significant concerns in this regard (119). A recent meta-analysis concluded there as insufficient evidence to support recommendation of its use (120)
Indications for therapy in adults with XLH are less clear. The occurrence of intractable bone pain and refractory non-union fractures often respond to treatment with calcitriol and phosphorus (121). However, data remain unclear regarding the effects of treatment on fracture incidence (which may not be increased in untreated patients). There does not appear to be any effect of this therapy on enthesopathy, however superior dentition appears to occur in the setting of higher medication exposure through adulthood as well as the entire life span (64). Muscle weakness and general well-being may occur with therapy in some adults. In sum, the decision to treat affected adults must be individualized. In general, it is beneficial to offer adults with significant symptomatology a trial of this therapy, but only if routine biochemical monitoring can be performed. Several detailed strategies for the management of children and adults with XLH are available (122-124).
A more recent development has been a more directed approach to the etiology of the renal phosphate loss. After demonstration of the efficacy of this strategy in the Hyp mouse model of XLH (40), trials of an antibody to the human FGF23 protein, burosumab (KRN23) have been conducted in children and adults (125-131) leading to its approval for use in both North America, several S. American countries, Europe, and other regions. The initial study using burosumab to treat in children with XLH resulted in improvement of radiographic features of rickets in concert with correction of abnormal biochemical indices after previous treated with conventional phosphate and active vitamin D therapy (127). Steady and stable correction of hypophosphatemia was attained with administration of the antibody every 2 weeks and a favorable safety profile was evident. The improved musculoskeletal status has been demonstrated to persist as seen in follow up extension studies for a total of 3 years (132). Moreover, one study has provided evidence that burosumab was superior to conventional therapy with calcitriol and phosphate in terms of skeletal improvement and growth (133).
AUTOSOMAL DOMINANT HYPOPHOSPHATEMIC RICKETS (ADHR)
Several studies have documented autosomal dominant inheritance of a hypophosphatemic disorder similar to XLH (134, 135). The phenotypic manifestations of this disorder include the expected hypophosphatemia due to renal phosphate wasting, lower extremity deformities, and rickets/osteomalacia. Affected patients also demonstrate normal serum 25(OH)D levels, while maintaining inappropriately normal serum concentrations of 1,25(OH)2D, in the presence of hypophosphatemia, all hallmarks of XLH (Table 2). PTH levels are normal. Long-term studies indicate that a few of the affected female patients demonstrate delayed penetrance of clinically apparent disease and an increased tendency for bone fracture, uncommon occurrences in XLH. In addition, among patients with the expected biochemical features documented in childhood, rare individuals lose the renal phosphate-wasting defect after puberty. As noted above, specific mutations in FGF23 in the 176-179 amino acid residue sequence are present in patients with ADHR (97). These mutations disrupt an RXXR furin protease recognition site, and the resultant mutant molecule is thereby protected from proteolysis, and resultant elevated circulating levels of FGF23 are the likely cause of the renal Pi wasting. Interestingly, circulating FGF23 levels can vary and reflect the activity of disease status (136).
Exploration of the waxing/waning severity of disease in ADHR has identified that iron may play a significant role in the regulation of circulating FGF23 (137). Iron deficiency appears to upregulate FGF23 expression, and in normal individuals, processing of the intact protein to its inactive N- and C-terminal fragments is efficient, thereby compensating for the increased intact FGF23 production seen with iron deficiency. Thus. in normal individuals who become iron deficient normal circulating levels of intact FGF23 are maintained despite the increase in production. However, in ADHR, inefficient processing of FGF23, due to the lack of protease recognition at the usual amino acid 179/180 cleavage site, may not be able to compensate for increased FGF23 synthesis during periods of iron deficiency. Thus, the waxing and waning clinical severity observed in some cases of ADHR may be amenable to iron supplementation, and provide a straightforward approach to therapy. A recently reported case demonstrates that correction of serum iron levels to high normal levels allowed for discontinuation of conventional rickets medications (138)
An apparent forme fruste of ADHR (autosomal dominant) hypophosphatemic bone disease has many of the characteristics of XLH and ADHR, but recent reports indicate that affected children display no evidence of rachitic disease. Because this syndrome is described in only a few small kindreds, and radiographically evident rickets is not universal in children with familial hypophosphatemia, these families may have ADHR. Further observations are necessary to discriminate this possibility.
AUTOSOMAL RECESSIVE HYPOPHOSPHATEMIC RICKETS (ARHR)
Families with phosphate wasting rickets inherited in an autosomal recessive manner have been described and demonstrate the same constellation of progressive rachitic deformities seen in both XLH and ADHR (98, 139). Moreover, the biochemical phenotype is manifest by the same measures of hypophosphatemia, excess urinary Pi losses, and aberrant vitamin D metabolism (normal circulating 25-OHD and 1,25(OH)2D levels, despite ambient hypophosphatemia) as observed in both XLH and ADHR. In addition to the expected phenotypic features, and in contrast to XLH, spinal radiographs of patients with ARHR reveal noticeably sclerotic vertebral bodies. In addition to the enlarged pulp chamber characteristic of teeth in individuals with XLH, enamel hypoplasia can be evident in heterozygotes. Of particular interest is the identification of elevated levels of FGF23 in the affected individuals. Experience with long-term follow-up is not widespread in ARHR and therapeutic response or guidelines have not been definitively established.
The identification of a progressive mineralization defect associated with hypophosphatemia in DMP1 knockout mice led to the consideration of homozygous loss of function in this candidate gene as a cause of ARHR. Indeed, this was proven to be the case for the first families identified with the disorder. Thus, the role of the osteocyte product, DMP1, appears as either part of the PHEX-FGF23 pathway, or at least can affect circulating FGF23 levels, perhaps independently of PHEX. These observations reinforce the central role that the osteocyte plays in mineral homeostasis.
Moreover, hypophosphatemic rickets in association with renal Pi wasting has been recently described in the setting of the extremely rare disorder, generalized arterial calcification of infancy (GACI) (99-101). This disorder occurs with homozygous loss-of-function mutations of ectonucleotide pyrophosphatase/phosphodiesterase-1 (ENPP1). Loss-of-function of ENPP1 results in the inability to generate the mineralization inhibitor, pyrophosphate, thereby disrupting the restriction of heterotopic (e.g., vascular) mineralization. GACI is often fatal, but hypophosphatemia, identified in the setting of elevated FGF23 levels in an adult with a homozygous ENPP1 mutation raised this consideration of rickets in survivors of GACI (140). Moreover, the patient’s son was affected with both GACI and hypophosphatemia. The mechanism by which this enzyme influences renal tubular phosphate wasting is not evident, and further study is necessary to understand this intriguing problem. One speculated mechanism may reflect a bone cell response to a relatively hypermineralized (or high-phosphate/low pyrophosphate) milieu which results in a compensatory, prolonged secretion of FGF23. Such a mechanism may effectively signal the kidney to reduce the body’s mineral load, but apparently cannot be down-regulated to protect against excessive Pi losses. Although there has been concern that the treatment of rickets in patients affected with GACI patients may promote worsening of vascular calcification, no evidence to sustain this concern has emerged and one long term observational report suggests that treatment does not worsen this finding (141). A recent phenotyping study with long-term observations in this regard corroborates this initial impression (102).
TUMOR-INDUCED OSTEOMALACIA
Rickets and/or osteomalacia have been associated with various types of tumors (96). In many cases, the metabolic disturbances improved or completely disappeared upon removal of the tumor, indicating a causal role of the tumor. Affected patients generally present with bone and muscle pain, muscle weakness, rickets/osteomalacia, and occasionally recurrent fractures of long bones. Biochemistries include hypophosphatemia secondary to renal phosphate wasting and normal serum levels of calcium and 25(OH)D. Serum 1,25(OH)2D is often overtly low or is otherwise inappropriately normal in the setting of hypophosphatemia (Table 2). Aminoaciduria and/or glucosuria may be present. Radiographic abnormalities include generalized osteopenia, pseudofractures and coarsened trabeculae, as well as widened epiphyseal plates in children. The histologic appearance of trabecular bone in affected subjects most often reflects the presence of a low turnover osteomalacia.
The large majority of patients with this syndrome harbor tumors of mesenchymal origin, including primitive-appearing, mixed connective tissue lesions. These tumors are often classified as osteoblastomas, osteochondromas, non-ossifying fibromas and ossifying fibromas. In addition, tumors of epidermal and endodermal derivation have been implicated as causal of the disease. Indeed, the observation of tumor-induced osteomalacia concurrent with breast carcinoma, prostate carcinoma oat cell carcinoma, small cell carcinoma, multiple myeloma and chronic lymphocytic leukemia have been reported.
Although this syndrome is relatively rare compared to XLH, investigation of causative tumors eventually led to the identification and isolation of FGF23 (38, 142), the mediator of many heritable hypophosphatemic disorders, and the recognition that this protein is the central factor in a major regulatory system affecting Pi homeostasis. Moreover, the discovery represented the first disorder related to the endocrine subfamily of FGFs, acting at distant sites with specificity of site activity conferred by the family of klotho co-receptors.
Regardless of the tumor cell type, the lesions at fault for the syndrome are often small, difficult to locate and present in obscure areas which include the nasopharynx, jaw, sinuses, the popliteal region and the suprapatellar area. In any case, a careful and thorough examination is necessary to document/exclude the presence of such a tumor. Indeed, CT and/or MRI scan of a clinically suspicious area should be undertaken. Recently newer imaging techniques such as octreotide scintigraphy or PET scans have been used to successfully identify tumors that remained unidentified by other means of localization. Newer agents with greater specificity for somatostatin receptors type 2 and type 5 appear to increase the sensitivity of PET scanning (143, 144), and co-registry with high resolution anatomic imaging has considerably advanced detection of small tumors.
Selective venous sampling has been suggested as a complementary approach to diagnosis. This technique may provide confirmation of local FGF23 secretion in suspicious areas identified by imaging (as to avoid unnecessary operations from false-positive imaging studies). The technique may serve to direct local imaging to anatomic regions defined by step-ups in FGF23 concentrations, but is limited by the relatively long half-life of FGF23, which may be misleading if the sampling is not in very close proximity to the offending tumor. Although useful in the settings mentioned above, the technique is not thought to be an optimal first-line approach in identification of TIO causing tumors (145).
Pathophysiology
TIO is a result of Pi wasting secondary to circulating factor(s) secreted by causal tumors. FGF23 has proven to be the primary factor identified in most patients where examination of serum levels or tumor material has occurred. Nevertheless, a variety of other factors have been considered as a potential part of the cascade that can lead to renal Pi wasting including: 1) FRP4 (frizzled related protein 4) (45), a secreted protein with phosphaturic properties, 2) FGF7, a paracrine FGF identified in TIO tumors that has been shown to directly inhibit renal Pi transport (47), 3) the SIBLING protein, MEPE (matrix extracellular phosphglycoprotein), which has been reported to generate fragments (ASARM peptide) with potential Pi wasting activity (44), 4) the SIBLING protein, DMP1, which has now been implicated in ARHR, and has been shown to be in particularly high abundance in TIO tumors (38, 98, 140, 146), and 5) the high molecular weight isoform of FGF2 (another paracrine FGF), which when expressed transgenically in mice, results in hypophosphatemic rickets (147). It is also possible that these or other tumor products may have direct effects on the mineralization function of the skeleton.
A novel genetic mechanism by which TIO tumors may develop autonomous FGF23 production involves a somatic chromosomal rearrangement which has been identified in a high proportion of TIO tumors (142). The rearrangement sequence predicts a fusion protein consisting of the N-terminal portion of fibronectin and the FGFR1 receptor. The extracellular fibronectin domain is proposed to promote dimerization and activation of the complex leading to downstream signaling resulting in FGF23 secretion. FGF23 itself is further proposed as an additive stimulus, amplifying FGF23 production as part of a feed-forward loop resulting in the substantial FGF23 production characteristic of TIO (148). More recently another rearrangement generating a fibronectin/FGF1 fusion protein has been described (149).
In contrast to these observations, other rare patients with TIO secondary to hematogenous malignancy manifest abnormalities that would suggest a different pathophysiologic mechanism. In these subjects a nephropathy induced with light chain proteinuria or other immunoglobulin derivatives appears to result in decreased renal tubular reabsorption of phosphate. Thus, light-chain nephropathy has been considered a possible mechanism for the TIO syndrome.
Treatment
The first and foremost treatment of TIO is complete resection of the tumor. However, recurrence of mesenchymal tumors, such as giant cell tumors of bone, or inability to resect completely certain malignancies, such as prostatic carcinoma, has resulted in development of alternative therapeutic intervention for the syndrome. In this regard, administration of 1,25(OH)2D alone or in combination with phosphorus supplementation has served as effective therapy for TIO. Doses of calcitriol required range from 1.5-3.0 µg/d, while those of phosphorus are 2-4 g/d. Although little information is available regarding the long-term consequences of such treatment, the high doses of medicine required raise the possibility that nephrolithiasis, nephrocalcinosis, and hypercalcemia may frequently complicate the therapeutic course. Indeed, hypercalcemia secondary to parathyroid hyperfunction has been documented in several subjects. Generally, these patients receive phosphorus as part of a combination regimen, exacerbating the path to parathyroid autonomy. Thus, as with treatment of XLH, careful assessment of parathyroid function, serum and urinary calcium, and renal function are essential to ensure safe and efficacious therapy. Recent studies using burosumab, the antiFGF23 antibody, have demonstrated improvement in both biochemical indices and biopsy parameters of osteomalacia in inoperable TIO (150).
In widespread fibrous dysplasia of bone (due to mosaic activating mutations in GNAS), neurofibromatosis and cutaneous skeletal hypophosphatemic syndrome (associated with somatic mutations in HRAS and NRAS) (151), hypophosphatemic osteomalacia/rickets can result as a result of elevated circulating FGF23 levels (152). Indeed, variable degrees of decreased renal tubular phosphate reabsorption, as assessed by TMP/GFR assessments, occur in patients with fibrous dysplasia of bone. Other primary skeletal disorders in which elevated FGF23 levels have been reported include osteoglophonic dysplasia (due to mutations in the FGFR1 receptor) (153), Jansen metaphyseal chondrodysplasia, (due to activating mutations of the PTH1 receptor) (154), opsismodysplasia (155), and in FAM20C mutations (110). The mechanism(s) by which elevations in FGF23 occur in these settings is not certain at this time.
Clinical Disorders: FGF23-independent Hypophosphatemia
HEREDITARY HYPOPHOSPHATEMIC RICKETS WITH HYPERCALCIURIA (HHRH)
This rare autosomal recessive disease is marked by hypophosphatemic rickets with hypercalciuria (156). Initial symptoms of the disorder generally manifest between 6 months to 7 years of age and usually consist of bone pain and/or deformities of the lower extremities. Such deformities may include genu varum or genu valgum or anterior bowing of the femur and coxa vara. Additional disease features include short stature, and radiographic signs of rickets or osteopenia. In contrast to XLH, muscle weakness may be elicited as a presenting symptom.
Many of the distinguishing characteristics of HHRH stem from the fact that HHRH is not a disorder of FGF23-mediated hypophosphatemia. In fact, levels are often decreased compared to the normal population. Consequently, in contrast to the previously described disorders in which renal phosphate transport is limited, patients with HHRH exhibit increased 1,25(OH)2D production. The resultant elevated serum calcitriol levels enhance gastrointestinal calcium absorption, which in turn increases the filtered renal calcium load and inhibits PTH secretion. Collectively these events produce the hypercalciuria observed in affected patients (Table 2). Although initially not thought to be part of the syndrome, the propensity for kidney stones to occur has been reported in several patients.
In general, the severity of the bone mineralization defect correlates inversely with the prevailing serum Pi concentration. Relatives of patients with evident HHRH may exhibit an additional mode of disease expression (157). These subjects manifest hypercalciuria and hypophosphatemia, but the abnormalities are less marked and occur in the absence of discernible bone disease, which would suggest a mild phenotype in the heterozygous state with certain mutations.
After mutations in the candidate NaPi-IIa gene were excluded as causal to HHRH, the genetic defect was identified in NaPi-IIc (27, 158), previously thought to be of less importance than the type IIa transporter. As would be predicted by the isolated loss of function of a Pi transporter, reduced serum Pi and increased renal Pi losses occur, independent of FGF23 status. However, unlike the findings in XLH, Pi wasting does not coexist with limitations in 1,25(OH)2D production, and the system retains its capacity to increase 1,25(OH)2D levels in response to the ambient hypophosphatemia. Recently it has been suggested that specific mutations in NaPi-IIc may be associated with sodium wasting and potentially the tendency to form urinary tract stones (159).
Patients with HHRH have been treated successfully with high-dose phosphorus (1 to 2.5 g/day in five divided doses) alone. In response to therapy, bone pain disappears and muscular strength improves substantially. Moreover, the majority of treated subjects exhibit accelerated linear growth, and radiologic signs of rickets are completely absent within several months. Despite this favorable response, limited studies indicate that such treatment does not completely heal the associated osteomalacia. Indeed, there is no collective experience with long-term follow-up of this rare disorder, and the necessity and/or complications of long-term therapy are not well-established. Curiously an accompanying osteoporosis appears to occur in concert, a finding that is also quite different from the usual picture in XLH.
AUTOSOMAL RECESSIVE HYPOPHOSPHATEMIC RICKETS WITH FANCONI SYNDROME AND PHOSPHATURIA ASSOCIATED WITH INFANTILE HYPERCALEMIA OF INFANCY
Although SLC34A3 (Na-Pi2c) mutations were identified as the mutated gene in the specific disorder of HHRH, homozygous loss-of-function mutations in SLC34A1 (Na-Pi2a) occur in yet another syndrome of autosomal recessive hypophosphatemic rickets accompanied by a generalized renal tubular disorder consistent with Fanconi syndrome (160). This disorder appears to occur with less frequency than HHRH, however in a recent search for genetic causes of idiopathic infantile hypercalcemia (IIH), loss-of-function mutations in SLC34A1 have been identified (161). Rickets is not a prominent feature of this disorder, but rather hypercalcemia, hypercalciuria, nephrocalcinosis and renal phosphate wasting. Resultant elevations in circulating 1,25(OH)2D levels lead to the hypercalcemia. It is important to distinguish this cause of IIH from those attributable to defects in CYP24A1 (vitamin D 24-hydroxylase) as therapy in cases attributable to NaPi2a deficiency should respond to phosphate supplementation whereas restriction of dietary calcium and vitamin D are recommended in cases due to CYP24A1 mutations.
DENT'S DISEASE (X-LINKED RECESSIVE HYPOPHOSPHATEMIA; XLRH)
The initial description of X-linked recessive hypophosphatemic rickets involved a family in which males presented with rickets or osteomalacia, hypophosphatemia, and a reduced renal threshold for phosphate reabsorption. In contrast to patients with XLH, affected subjects exhibited hypercalciuria, elevated serum 1,25(OH)2D levels (Table 1), and proteinuria of up to 3 g/day. Patients also developed nephrolithiasis and nephrocalcinosis with progressive renal failure in early adulthood. Female carriers in the family were not hypophosphatemic and lacked any biochemical abnormalities other than hypercalciuria. Three related syndromes have been reported independently: X-linked recessive nephrolithiasis with renal failure, Dent's disease, and low-molecular-weight proteinuria with hypercalciuria and nephrocalcinosis. These syndromes differ in degree from each other, but common themes include proximal tubular reabsorptive failure, nephrolithiasis, nephrocalcinosis, progressive renal insufficiency, and, in some cases, rickets or osteomalacia. Identification of mutations in the voltage-gated chloride-channel gene CLCN5 in all four syndromes has established that they are phenotypic variants of a single disease and are not separate entities (162,163). However, the varied manifestations that may be associated with mutations in this gene, particularly the presence of hypophosphatemia and rickets/osteomalacia, underscore that environmental differences, diet, and/or modifying genetic backgrounds may influence phenotypic expression of the disease.
INTESTINAL MALABSORPTION OF PHOSPHATE
Although primary disorders of intestinal phosphate absorption have not been considered of clinical significance, we have encountered a curious phenomenon of phosphate malabsorption in children with complex disorders associated with intestinal compromise, when fed amino-acid based elemental formula (164,165). Associated tube-feeding and use of antacid medications appear to be risk factors, and the phenomenon does not appear to occur when used for the labeled indication of milk protein allergy in children who are otherwise healthy (166). We have recommended that serum phosphorus levels be monitored periodically with the use of such formulas.
REFERENCES
- Yanagawa N, Nakhoul F, Kurokawa K, Lee DBN. Physiology of phosphorus metabolism. In: Narins RG, ed. Clinical disorders of fluid and electrolyte metabolism. 5th ed. New York: McGraw Hill,1994; 307-371.
- Lee DBN, Walling MW, Brautbar N . Intestinal phosphate absorption: Influence of vitamin D and non-vitamin D factors. Am J Physiol. 1986; 250: G369-G373.
- Cross HS, Debiec H, Peterlik M. Mechanism and regulation of intestinal phosphate absorption. Miner Electrolyte Metab. 1990;16:115-124.
- Marks J, Debnam ES, Unwin RJ. The role of the gastrointestinal tract in phosphate homeostasis in health and chronic kidney disease. Curr Opin Nephrol Hypertens. 2013; 22:481-487.
- Sabbagh Y, O’Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, Schiavi SCIntestinal npt2b plays a major role in phosphate absorption homeostasis. J Am Soc Nephrol. 2009; 20:2348–2358.
- Debiec H, Lorenc R. Identification of Na+,Pi-binding protein in kidney and intestinal brush-border membranes. Biochem J. 1988; 225:185-191.
- Katai K, Miyamoto K, Kishida S, Segawa H, Nii T, Tankaka H, Tani Y, Arai H, Tatsumi S, Morita K, Taketani Y, Takeda E. Regulation of intestinal Na+-dependent phosphate co-transporters by a low phosphate diet and 1,25-dihydroxyvitamin D3. Biochem J. 1999; 3:705-712.
- Hilfiker H, Hattenhauer O, Traebert M, Forster I, Murer H, Biber J. Characterization of a murine type II sodium-phosphate cotransporter expressed in mammalian small intestine. Proc Natl Acad Sci USA. 1998; 95:14564-14569.
- Bai L, Collins JF, Ghishan FK. Cloning and characterization of a type III Na-dependent phosphate cotransporter from mouse intestine. Am J Physiol Cell Physiol. 2000; 279:C1135-C1143.
- Xu H, Bai L, Collins JF, Ghishan FK. Age-dependent regulation of rat intestinal type IIb sodium-phosphate cotransporter by 1,25-(OH)2 vitamin D3. Am J Physiol Cell Physiol. 2002; 282:C487-C493.
- Marks J, Debnam ES, Unwin RJ. Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol. 2009; 299:F285-F296.
- Candeal E, Caldas YA, Guillén N, Levi M, Sorribas V. Intestinal phosphate absorption is mediated by multiple transport systems in rats. Am J Physiol Gastrointest Liver Physiol. 2017; 312(4): G355-G366.
- Mizgala CL, Quamme GA. Renal handling of phosphate. Physiol Rev. 1985; 65:431-466.
- Harris CA, Sutton RA, Dirks JH. Effects of hypercalcemia on tubular calcium and phosphate ultrafilterability and tubular reabsorption in the rat. Am J Physiol. 1977; 233:F201-F206.
- Knox FG, Haramati A. Renal regulation of phosphate excretion. In: Seldin DW, Giebisch G, eds. The Kidney: Physiology and Pathophysiology. New York: Raven Press; 1981; 1381.
- Berndt TJ, Knox FG. Proximal tubule site of inhibition of phosphate reabsorption by calcitonin. Am J Physiol. 1984; 246:F927-F930.
- Legati A, Giovannini D, Nicolas G, Lopez-Sanchez U, Quintans B, Oliveira JR, Sears RL, Ramos EM, Spiteri E, Sobrido MJ, Carracedo A, Castro-Fernandez C, Cubizolle S, Fogel BL, Goizet C, Jen JC, Kirdlarp S, Lang AE, Miedzybrodzka Z, Mitarnun W, Paucar M, Paulson H, Pariente J, Richard AC, Salins NS, Simpson SA, Striano P, Svenningsson P, Tison F, Unni VK, Vanakker O, Wessels MW, Wetchaphanphesat S, Yang M, Boller F, Campion D, Hannequin D, Sitbon M, Geschwind DH, Battini JL, Coppola G. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet. 2015; 47(6):579–581.
- Ansermet C, Moor MB, Centeno G, Auberson M, Hu DZ, Baron R, Nikolaeva S, Haenzi B, Katanaeva N, Gautschi I, Katanaev V, Rotman S, Koesters R, Schild L, Pradervand S, Bonny O, Firsov D. Renal fanconi syndrome and hypophosphatemic rickets in the absence of xenotropic and polytropic retroviral receptor in the nephron. J Am Soc Nephrol. 2017; 28(4):1073-1078.
- Schwab SJ, Klahr S, Hammerman MR.Na+ gradient-dependent Pi uptake in basolateral membrane vesicles from dog kidney. Am J Physiol. 1984; 246: F633-F639.
- Villa-Bellosta R, Ravera S, Sorribas V, Stange G, Levi M, Murer H, Biber J, Forster IC. The Na+-Pi cotransporter PiT-2 (SLC20A2) is expressed in the apical membrane of rat renal proximal tubules and regulated by dietary Pi. Am J Physiol Renal Physiol. 2009; 296(4): F691-F699.
- Murer H, Forster I, Hernando N, Lambert G, Traebert M, Biber J. Post-transcriptional regulation of the proximal tubule Na+-phosphate transporter type II in response to PTH and dietary phosphate. Am J Physiol Renal Physiol. 1999; 277: F676-F684.
- Murer H, Forster I, Hilfiker H, Pfister M, Kaissling B, Lotscher M, Biber J. Cellular/molecular control of renal Na+/Pi cotransport. Kidney Int. 1988; 65:S2-S10.
- Bacconi A, Virkki LV, Biber J, Murer H, Forster IC. Renouncing electroneutrality is not free of charge: switching on electrogenicity in a Na+-coupled phosphate cotransporter. Proc Natl Acad Sci USA. 2005; 102(35):12606-12611.
- Oberbauer R, Schreiner GF, Biber J, Murer H, Meyer TW.In vivo suppression of the renal Na+/Pi cotransporter by antisense oligonucleotides. Proc Natl Acad Sci USA. 1996; 93:4903-4906.
- Beck I, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt 2 in mice leads to severe renal phosphate wasting, hypercalciuria and skeletal annomalies. Proc Natl Acad Sci USA. 1998; 95:5372-5377.
- Hoag HH, Gauthier C, Martel I, Tenenhouse HS. Effects of Npt2 gene ablation and low Pi-diet on renal Na+-phosphate cotransport and cotransporter gene expression. J Clin Invest. 1999; 104:679-686.
- Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabedian M, Sermet I, Fujiwara TM, Morgan KN, Tenenhouse HS, Jüppner H. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Gen. 2006; 78:179-192.
- Biber J, Hernando N, Forster I. Phosphate transporters and their function. Annu Rev Physiol. 2013; 75:535-550.
- Lötscher M, Scarpetta Y, Levi M, Halaihel N, Wang H, Zajicek HK, Biber J, Murer H, Kaissling B. Rapid downregulation of rat renal Na/P(i) cotransporter in response to parathyroid hormone involves microtubule rearrangement. J Clin Invest. 1999; 104(4):483-494.
- Wagner CA, Hernando N, Forster IC, Biber J. The SLC34 family of sodium-dependent phosphate transporters. Pflugers Arch. 2014; 466:139-153.
- Segawa H, Yamanaka S, Ito M, Kuwahata M, Shono M, Yamamoto T, Miyamoto K. Internalization of renal type IIc Na-Pi cotransporter in response to a high-phosphate diet. Am J Physiol Renal Physiol. 2005; 288:F587-F596.
- Foster IC, Hernando N, Biber J, Murer H. Proximal tubular handling of phosphate: a molecular perspective. Kidney Int. 2006; 70:1548-1559.
- Farrow EG, White KE. Recent advances in renal phosphate handling. Nat Rev Nephrol. 2010; 6(4):207-217.
- Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006; 91:3144-3149.
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006; 444:770-774.
- Belov AA, Mohammadi M. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb Perspect Biol. 2013; 5:6.
- Andrukhova O, Zeitz U, Goetz R, Mohammadi M, Lanske B, Erben RG. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone. 2012; 51:621-628.
- Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T . Cloning and characterization of FGF-23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. 2001; 98:6500-6505.
- Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J. 2006; 6:720-722.
- Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H, Urakawa I, Fujita T, Wada M, Yamashita T, Fukumoto S, Shimada T. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2009 Nov;24(11):1879-88.
- Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Shawkat Razzaque M, Rosenblatt KP, Baum MG, Kuro-O M, Moe OW. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010; 24(9):3438-3450.
- Brownstein CA, Adler F, Nelson-Williams C, Iijma J, Imura A, Nabehsima Y, Carpenter TO, Lifton RP. A translocation causing increased α–Klotho level results in hypophosphatemic rickets and hyperparathyroidism. PNAS USA. 2008; 105:3455-3460.
- Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, Li J, Shehadeh LA, Hare JM, David V, Martin A, Fornoni A, Di Marco GS, Kentrup D, Reuter S, Mayer AB, Pavenstädt H, Stypmann J, Kuhn C, Hille S, Frey N, Leifheit-Nestler M, Richter B, Haffner D, Abraham R, Bange J, Sperl B, Ullrich A, Brand M, Wolf M, Faul C. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015; 22(6):1020-32.
- Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MH, Oudet CL. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics. 2000; 67:54-68.
- Berndt T, Craig TA, Howe AE, Vassiliadis J, Reczek D, Finnegan R, Jan de Beur SM, Schiavi SC, Kumar R.Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003; 112:785-794.
- Wagner GF, Dimattia GE. The stanniocalcin family of proteins. J Exp Zoolog, A Comp Exp Biol. 2006; 305:769-780.
- Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. FGF7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab. 2005; 90:1012-1020.
- Dallas SL, Prideaux M, Bonewald LF.The osteocyte: an endocrine cell ... and more. Endocr Rev. 2013; 34:658-690.
- Murali SK, Andrukhova O, Clinkenbeard EL, White KE, Erben RG. Excessive osteocytic Fgf23 secretion contributes to pyrophosphate accumulation and mineralization defect in Hyp mice. PLoS Biol. 2016;14(4):e1002427.
- Hoac B, Østergaard M, Wittig NK, Boukpessi T, Buss DJ, Chaussain C, Birkedal H, Murshed M, McKee MD. Genetic ablation of osteopontin in osteomalacic Hyp mice partially rescues the deficient mineralization without correcting hypophosphatemia. J Bone Miner Res. 2020;35(10):2032-2048.
- Bai X, Miao D, Goltzman D, Karaplis AC. Early lethality in hyp mice with targeted deletion of Pth gene.Endocrinology. 2007; 148(10):4974-83.
- Carpenter TO, Olear EA, Zhang JH, Ellis BK, Simpson CA, Cheng D, Gundberg CM, Insogna KL. Effect of paricalcitol on circulating parathyroid hormone in X-linked hypophosphatemia: a randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab. 2014; 99(9):3103-11.
- Lanske B, Razzaque MS. Molecular interactions of FGF23 and PTH in phosphate regulation. Kidney Int. 2014; 86(6):1072-4.
- Sabbagh Y, Carpenter TO, Demay M. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Nat Acad Sci. 2005; 102:9637-9642.
- Glimcher MJ .In: Aurbach GD ed. Handbook of physiology: endocrinology, parathyroid gland, sect 7, vol 7. Washington, D.C.: American Physiological Society. 1996; 21-32.
- Bordier PJ, Tun Chot S. Quantitative histology of metabolic bone disease. Clin Endocrinol Metab. 1972; 1:197-215.
- Frame B, Parfitt AM. Osteomalacia: current concepts. Ann Intern Med. 1978; 89:966-982.
- Mumm S, Huskey M, Cajic A, Wollberg V, Zhang F, Madson KL, Wenkert D, McAlister WH, Gottesman GS, Whyte MP. PHEX 3'-UTR c.*231A>G near the polyadenylation signal is a relatively common, mild, American mutation that masquerades as sporadic or X-linked recessive hypophosphatemic rickets. J Bone Miner Res. 2015; 30(1):137-43.
- Harrison HE, Harrison HC, Lifshitz F, Johnson AD. Growth disturbance in hereditary hypophosphatemia. Am J Dis Child. 1996; 112:290-297.
- Mao M, Carpenter TO, Whyte MP, Skrinar A, Chen CY, San Martin J, Rogol AD. Growth curves for children with X-linked Hypophosphatemia. J Clin Endocrinol Metab 105: 32439, 2020.
- Williams TF, Winters RW. Familial (hereditary) vitamin D-resistant rickets with hypophosphatemia. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, eds. The metabolic basis of inherited disease. 3rd ed. New York: McGraw-Hill. 1983; 1465-1485.
- Tracey WE, Campbell RA. Dentofacial development in children with vitamin D resistant rickets. J Am Dent Assoc. 1968; 76:1026-1031.
- Shields ED, Scriver CR, Reade T, Fujiwara TM, Morgan K, Ciampi A, Schwartz S. X-linked hypophosphatemia: the mutant gene is expressed in teeth as well as in kidney. Am J Human Gen. 1990; 46:434-442.
- Marie PJ, Glorieux FH. Relation between hypomineralized periosteocytic lesions and bone mineralization in vitamin D-resistant rickets. Calcif Tissue Int. 1983; 35:443-448.
- Polisson RP, Martinex S, Khoury M, Harrell RM, Lyles KW, Friedman N, Harrelson JM, Reisner E, Drezner MK. Calcificantion of entheses associated with X-linked hypophosphatemic osteomalacia. N Engl J Med. 1985; 313:1-6.
- Pierce DS, Wallace WM, Herndon CH. Long term treatment of vitamin D-resistant rickets. J Bone Joint Surg Am. 1964; 46:978-997.
- Connor J, Olear EA, Insogna KL, Katz L, Baker SD, Kaur RD, Simpson CA, Sterpka J, Dubrow R, Zhang JH, Carpenter TO. Conventional therapy in adults with X-linked hypophosphatemia: effects on enthesopathy and dental disease. J Clin Endocrinol Metab. 2015; 100:3625-3632.
- Steindijk R. On the pathogenesis of vitamin D resistant rickets and primary vitamin D resistant rickets. Helv Paediatr Acta. 1962; 17:65-85.
- Stickler GB External calcium and phosphorus balances in vitamin D-resistant rickets. J Pediatr. 1963; 63:942-948.
- Drezner MK, Lyles KW, Haussler MR, Harrelson JM. Evaluation of a role for 1,25-dihydroxyvitamin D3 in the pathogenesis and treatment of X-linked hypophosphatemic rickets and osteomalacia. J Clin Invest. 1980; 66:1020-1032.
- Haddad JG, Chyu KJ, Hahn TJ, Stamp TCB. Serum concentrations of 25-hydroxyvitamin D in sex linked hypophosphatemic vitamin D-resistant rickets. J Lab Clin Med. 1973; 81:22-27.
- Tenenhouse HS. Abnormal renal mitochondrial 25-hydroxyvitamin D3-1-hydroxylase activity in the vitamin D and calcium deficient X-linked Hyp mouse. Endocrinology. 1983; 113:816-818.
- Roy S, Martel J, Ma S, Tenenhouse HS. Increased renal 25-hydroxyvitamin D3-24-hydroxylase messenger ribonucleic acid and immunoreactive protein in phosphate-deprived Hyp mice: a mechanism for accelerated 1,25-dihydroxyvitamin D3 catabolism in X-linked hypophosphatemic rickets. Endocrinology. 1994; 134:1761-1767.
- Winters RW, Graham JB, Williams TF, McFalls VW, Burnett CH. A genetic study of familial hypophosphatemia and vitamin D-resistant rickets with a review of the literature. Medicine (Baltimore). 1958; 37:97-142.
- Burnett CH, Dent CE, Harper C, Warland BJ Vitamin D resistant rickets: analysis of 24 pedigrees and hereditary and sporadic cases. Am J Med. 1964; 36:222-232.
- Francis F, Henning S, Korn B, Reinhardt R, de Jong P, Poustka A, Lehrach H, Rowe PSN, Goulding JN, Summerfield T, Mountford R, Read AP, Popowska E, Pronicka E, Davies KE, O’Riordan JLH, Econs MJ, Nesbitt T, Drezner MK, Oudet C, Hanauer A, Strom TM, Meindl A, Lorenz B, Cagnoli M, Mohnike KL, Murken J, Meitinger T. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet. 1995; 11:130-136.
- Lipman ML, Dibyendu P, Hugh PJ, Bennett JE, Henderson ES, Yingnian S, Goltzman D, Daraplis AC. Cloning of human Pex cDNA: expression subcellular localization and endopeptidase activity. J Biol Chem. 1998; 273:13729-13737.
- Thompson DL, Roche PC, Drezner MK, Salisbury JL, Sabbagh Y, Tenenhouse HS, Grande JP, Poeschlia EM, Kumar R.Ontogeny of PHEX/PEX expression in the mouse embryo and studies on the subcellular localization of PHEX/PEX in osteoblasts. J Bone Miner Res. 2002; 17:311-320.
- Sabbagh, Y., Boileau, G., Campos, M., Carmona, A. K., & Tenenhouse, H. S. (2003). Structure and function of disease-causing missense mutations in the PHEX gene. TheJournal of Clinical Endocrinology and Metabolism, 88(5), 2213– 2222.
- Sabbagh, Y., Boileau, G., DesGroseillers, L., & Tenenhouse, H. S. (2001). Disease-causing missense mutations in the PHEX gene interfere with membrane targeting of the recombinant protein. Human Molecular Genetics, 10(15), 1539–1546.
- Beck L, Soumounou Y, Martel J, Krishnamurthy G, Gauthier C, Goodyer CG, Tenenhouse HS. Pex/PEX tissue distribution and evidence for a deletion in the 3' region of the Pex gene in X-linked hypophosphatemic mice. J Clin Invest. 1997; 99:1200-1209.
- Zoidis E, Zapf J, Schmid C. Phex cDNA cloning from rat bone and studies on phex mRNA expression: tissue-specificity, age-dependency, and regulation by insulin-like growth factor (IGF) I in vivo. Mol Cell Endocrinol. 2000; 168:41-51.
- Ruchon AF, Tenenhouse HS, Marcinkiewicz M, Siegfried G, Aubin JE, DesGroseillers L, Crine P, Boileau G. Developmental expression and tissue distribution of Phex protein: effect of the Hyp mutation and relationship to bone markers. J Bone Miner Res. 2000; 15:1440-1450.
- Ruchon AF, Marcinkiewicz M, Siegfried G, Tenenhouse HS, DesGroseillers L, Crine P, Boileau G. Pex mRNA is localized in developing mouse osteoblasts and odontoblasts. J Histochem Cytochem. 1998; 46:459-468.
- Sarafrazi S, Daugherty SC, Miller N, Boada P, Carpenter TO, Chunn L, Dill K, Econs MJ, Eisenbeis S, Imel EA, Johnson B, Kiel MJ, Krolczk S, Ramesan P, Truty R, Sabbagh Y. Novel PHEX gene locus-specific database: Comprehensive characterization of vast number of variants associated with X-Linked Hypophosphatemia (XLH). HumanMutation 43(2):143-157, 2022.
- Liu S, Guo R, Tu Q, Quarles LD. Overexpression of phex in osteoblasts fails to rescue the hyp-mouse phenotype. J Biol Chem. 2002; 277:3686-3697.
- Bai X, Miao D, Panda D, Grady S, McKee MD, Goltzman D, Karaplis AC. Partial rescue of the hyp phenotype by osteoblast-targeted PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) expression. Mol Endocrinol. 2002; 16:2913-2925.
- Boskey A, Frank A, Fujimoto Y, Spevak L, Verdelis K, Ellis B, Philbrick W, Carpenter T. The PHEX transgene corrects mineralization defects in 9-month-old hypophosphatemic mice. Calcif Tiss Int. 2009; 84:126-137.
- David V, Martin A, Hedge AM, Drezner MK, Rowe PS. ASARM peptides: PHEX-dependent and -independent regulation of serum phosphate. Am J Physiol Renal Physiol. 2011; 300(3):F783-91.
- Morgan JM, Hawley WL, Chenoweth AI, Retan WJ, Diethelm AG. Renal transplantation in hypophosphatemia with vitamin D-resistant rickets. Arch Intern Med. 1974; 134(3):549-52
- Meyer RA Jr, Meyer MH, Gray RW . Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J Bone Miner Res. 1989; 4:493-500.
- Nesbitt T, Coffman TM, Griffiths R, Drezner MK. Cross transplantation of kidneys in normal and hyp-mice: evidence that the hyp-mouse phenotype is unrelated to an intrinsic renal defect. J Clin Invest. 1992; 89:1453-1459.
- Tenenhouse HS, Beck L. Renal Na+-P cotransporter gene expression in X-linked Hyp and Gy mice. Kidney Int. 1996; 49:1027-1032.
- Tenenhouse HS, Martel J, Biber J, Murer H. Effect of P(i) restriction on renal Na(+)-P(i) cotransporter mRNA and immunoreactive protein in X-linked Hyp mice. Am J Physiol. 1995; 268: F1062-F1069.
- Tenenhouse HS, Martel J, Gauthier C, Segawa H, Miyamoto K. Differential effects of Npt2a gene ablation and X-linked Hyp mutation on renal expression of Npt2c. Am J Physiol Renal Physiol. 2003; 285(6):F1271-F1278.
- White KE, Jonsson KB, Carn G, Hampson G, Spector TD, Mannstadt M, Lorenz-Depiereux B, Miyauchi A, Yang IM, Ljunggren O, Meitinger T, Strom TM, Jüppner H, Econs MJ. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001; 86:497-500.
- The ADHR Consortium. Autosomal dominant hypophosphatemic rickets is associated with mutations in FGF-23. Nat Genet. 2000; 26:345-348.
- Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006; 38:1310-1315.
- Levy-Litan V, Hershkovitz E, Avizov L, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010; 86(2):273-278..
- Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. 2010; 86(2):267-272.
- Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nürnberg P. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003; 34(4):379-381.
- Ferreira CR, Hackbarth ME, Ziegler SG, Pan KS, Roberts MS, Rosing DR, Whelpley MS, Bryant JC, Macnamara EF, Wang S, Müller K, Hartley IR, Chew EY, Corden TE, Jacobsen CM, Holm IA, Rutsch F, Dikoglu E, Chen MY, Mughal MZ, Levine MA, Gafni RI, Gahl WA. Prospective phenotyping of long-term survivors of generalized arterial calcification of infancy (GACI). Genet Med. 2021; 23(2):396-407.
- Yoskida T, Fujimori T, Nabeshima Y. Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1 -hydroxylase gene. Endocrinology. 2002; 143:683-689.
- Brownstein C, Zhang J, Stillman A, Ellis B, Troiano N, Adams DJ, Gundberg CM, Lifton RP, Carpenter TO. Increased bone volume and correction of HYP mouse hypophosphatemia in the Klotho/HYP mouse. Endocrinology. 2010; 151: 492-501.
- Smith RC, O'Bryan LM, Farrow EG, Summers LJ, Clinkenbeard EL, Roberts JL, Cass TA, Saha J, Broderick C, Ma YL, Zeng QQ, Kharitonenkov A, Wilson JM, Guo Q, Sun H, Allen MR, Burr DB, Breyer MD, White KE. Circulating αKlotho influences phosphate handling by controlling FGF23 production. J Clin Invest. 2012; 122:4710-4715.
- Ichikawa S, Sorenson AH, Austin AM, Mackenzie DS, Fritz TA, Moh A, Hui SL, Econs MJ. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology. 2009; 150(6):2543-2550.
- Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V, Zelikovic I, Raimer S, Metzker A, Richard G, Sprecher E. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004; 36:579-81.
- Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, Padgett LR, Austin AM, Sorenson AH, Pejin Z, Topouchian V, Quartier P, Cormier-Daire V, Dechaux M, Malandrinou FCh, Singhellakis PN, Le Merrer M, Econs MJ.Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A. 2010; 152A:896-903.
- Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, Koller A, Nizet V, White KE, Dixon JE. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A. 2014; 111:5520-5525.
- Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, Bjerknes R. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res. 2013; 28:1378-1385.
- Larsson T, Yu X, Davis SI, Draman MS, Mooney SD, Cullen MJ, White KE. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab. 2005; 90:2424-2427.
- Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007; 117(9):2684-2691.
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI.Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997; 390:45-51.
- Glorieux FH, Marie PJ, Pettifor JM, Delvin EE. Bone response to phosphate salts, ergocalciferol, and calcitriol in hypophosphatemic vitamin D-resistant rickets. N Engl J Med. 1980; 303:1223-1231.
- Costa T, Marie P, Scriver CR, Cole DEC, Reade TM, Norgrady B, Glorieux FH, Delvin EE. X-linked hypophosphatemia: effect of calcitriol on renal handling of phosphate, serum phosphate and bone mineralization. J Clin Endocrinol Metab. 1981; 52:463-477.
- Harrell RM, Lyles KW, Harrelson JM, Freedman NE, Drezner MK . Healing of bone disease in X-linked hypophosphatemic rickets/osteomalacia: induction and maintenance with phosphorus and calcitriol. J Clin Invest. 1985; 75:1858-1864.
- Baroncelli GI, Bertelloni S, Ceccarelli C, Saggese G 2001 Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets. J Pediatr. 1985; 138:236-243.
- Haffner D, Wuhl E, Blum WF, Schaefer F, Mehls O. Disproportionate growth following long-term growth hormone treatment in short children with X-linked hypophosphataemia. Eur J Pediatr. 1995; 154:610-613.
- Zivicnjak M, Schnabel D, Staude H, et al. Three-Year Growth Hormone Treatment in Short Children with X-Linked Hypophosphatemic Rickets: Effects on Linear Growth and Body Disproportion. J Clin Endocrinol Metab.2011; 96(12):E2097-E2105.
- Smith S, Remmington T.Recombinant growth hormone therapy for X-linked hypophosphatemia in children. Cochrane Database Syst Rev. 2021 Oct 7;10(10):CD004447.
- Sullivan W, Carpenter TO, Glorieux F, Travers R, Insogna K. A prospective trial of phosphate and 1,25-dihydroxyvitamin D3 therapy on symptomatic adults with X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 1992; 75:879-885.
- Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician's guide to X-linked hypophosphatemia. J Bone Miner Res. 2011; 26(7):1381-1388.
- Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, Wicart P, Bockenhauer D, Santos F, Levtchenko E, Harvengt P, Kirchhoff M, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenicky P, Rejnmark L, Linglart A. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019 Jul;15(7):435-455.
- Laurent MR, De Schepper J, Trouet D, Godefroid N, Boros E, Heinrichs C, Bravenboer B, Velkeniers B, Lammens J, Harvengt P, Cavalier E, Kaux JF, Lombet J, De Waele K, Verroken C, van Hoeck K, Mortier GR, Levtchenko E, Vande Walle J. Consensus Recommendations for the Diagnosis and Management of X-Linked Hypophosphatemia in Belgium. Front Endocrinol (Lausanne). 2021 Mar 19;12:641543. doi: 10.3389/fendo.2021.641543. eCollection 2021.
- Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, Kawakami T, Ito T, Zhang X, Humphrey J, Insogna KL, Peacock M.Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. 2014; 124:1587-1597.
- Imel EA, Zhang X, Ruppe MD, Weber TJ, Klausner MA, Ito T, Vergeire M, Humphrey JS, Glorieux FH, Portale AA, Insogna K, Peacock M, Carpenter TO. Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab. 2015; 100, 2565-2573.
- Carpenter TO, Whyte MP, Imel EA, Boot AM, Högler W, Linglart A, Padidela R, Van't Hoff W, Mao M, Chen CY, Skrinar A, Kakkis E, San Martin J, Portale AA. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N Engl J Med. 2018 May 24;378(21):1987-1998.
- Whyte MP, Carpenter TO, Gottesman GS, Mao M, Skrinar A, San Martin J, Imel EA. Efficacy and safety of burosumab in children aged 1-4 years with X-linked hypophosphataemia: a multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019 Mar;7(3):189-199.
- Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, Simmons JH, Padidela R, Namba N, Cheong HI, Pitukcheewanont P, Sochett E, Högler W, Muroya K, Tanaka H, Gottesman GS, Biggin A, Perwad F, Mao M, Chen CY, Skrinar A, San Martin J, Portale AA.Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet. 2019 Jun 15;393(10189):2416-2427.
- Insogna KL, Briot K, Imel EA, Kamenický P, Ruppe MD, Portale AA, Weber T, Pitukcheewanont P, Cheong HI, Jan de Beur S, Imanishi Y, Ito N, Lachmann RH, Tanaka H, Perwad F, Zhang L, Chen CY, Theodore-Oklota C, Mealiffe M, San Martin J, Carpenter TO; AXLES 1 Investigators. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults With X-Linked Hypophosphatemia: Week 24 Primary Analysis. J Bone Miner Res. 2018 Aug;33(8):1383-1393.
- Insogna KL, Rauch F, Kamenický P, Ito N, Kubota T, Nakamura A, Zhang L, Mealiffe M, San Martin J, Portale AA. Burosumab Improved Histomorphometric Measures of Osteomalacia in Adults with X-Linked Hypophosphatemia: A Phase 3, Single-Arm, International Trial. J Bone Miner Res. 2019 Dec;34(12):2183-2191.
- Linglart A, Imel EA, Whyte MP, Portale AA, Högler W, Boot AM, Padidela R, Van't Hoff W, Gottesman GS, Chen A, Skrinar A, Scott Roberts M, Carpenter TO. Sustained Efficacy and Safety of Burosumab, a Monoclonal Antibody to FGF23, in Children With X-Linked Hypophosphatemia. J Clin Endocrinol Metab. 2022 Feb 17;107(3):813-824.
- Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, Simmons JH, Padidela R, Namba N, Cheong HI, Pitukcheewanont P, Sochett E, Högler W, Muroya K, Tanaka H, Gottesman GS, Biggin A, Perwad F, Mao M, Chen CY, Skrinar A, San Martin J, Portale AA. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet. 2019 Jun 15;393(10189):2416-2427.
- Harrison HE, Harrison HC. Rickets and osteomalacia. In: disorders of calcium and phosphate metabolism in childhood and adolescence. Philadelphia: WB Saunders. 1979; 141-256.
- Econs M, McEnery P. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate wasting disorder. J Clin Endocrinol Metab. 1997; 82:674-681.
- Imel EA, Hui SL, Econs MJ. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J Bone Miner Res. 2007; 22(4):520-526.
- Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab. 2011; 96: 3541-3549.
- Kapelari K, Köhle J, Kotzot D, Högler W. Iron supplementation associated with loss of phenotype in autosomal dominant hypophosphatemic rickets. J Clin Endocrinol Metab. 2015; 100(9):3388-92.
- Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Jüppner H, Strom TM. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006; 38:1248-1250..
- Rutsch F, Böyer P, Nitschke Y, Ruf N, Lorenz-Depierieux B, Wittkampf T, Weissen-Plenz G, Fischer RJ, Mughal Z, Gregory JW, Davies JH, Loirat C, Strom TM, Schnabel D, Nürnberg P, Terkeltaub R; GACI study group hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008; 1(2):133-140.
- Ferreira CR, Ziegler SG, Gupta A, Groden C, Hsu KS. Treatment of hypophosphatemic rickets in generalized arterial calcification of infancy (GACI) without worsening of vascular calcification. Am J Med Genet A. 2016; 170A(5):1308-11.
- Bowe AE, Finnegan R, Jan de Beur SM, Cho J, Levine MA, Kumar R, Schiavi SC. FGF-23 inhibits renal tubular P transport and is a PHEX substrate. Biochem Biophys Res Commun. 2001; 284:977-981.
- Breer S, Brunkhorst T, Beil FT, Peldschus K, Heiland M, Klutmann S, Barvencik F, Zustin J, Gratz KF, Amling M. 68Ga DOTA-TATE PET/CT allows tumor localization in patients with tumor-induced osteomalacia but negative 111In-octreotide SPECT/CT. Bone. 2014; 64:222-227.
- El-Maouche D, Sadowski SM, Papadakis GZ, Guthrie L, Cottle-Delisle C, Merkel R, Millo C, Chen CC, Kebebew E, Collins MT. 68Ga-DOTATATE for Tumor Localization in Tumor-Induced Osteomalacia. J Clin Endocrinol Metab. 2016 ;101(10):3575-3581.
- Andreopoulou P, Dumitrescu CE, Kelly MH, Brillante BA, Cutler Peck CM, Wodajo FM, Chang R, Collins MT. Selective venous catheterization for the localization of phosphaturic mesenchymal tumors. J Bone Miner Res. 2011; 26:1295-1302.
- Jan De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, Estes S, Manavalan P, Petroziello J, Madden SL, Cho JY, Kumar R, Levine MA, Schiavi SC. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res. 2002; 17:1102-1110.
- Xiao L, Naganawa T, Lorenzo J, Carpenter TO, CoffinJD, Hurley MM. Nuclear isoforms of fibroblast growth factor 2 are novel inducers of hypophosphatemia via modulation of FGF23 and Klotho. J Biol Chem. 2010; 285:2843-2846.
- Lee JC, Jeng YM, Su SY, Wu CT, Tsai KS, Lee CH, Lin CY, Carter JM, Huang JW, Chen SH, Shih SR, Mariño-Enríquez A, Chen CC, Folpe AL, Chang YL, Liang CW. Identification of a novel FN1-FGFR1 genetic fusion as a frequent event in phosphaturic mesenchymal tumour. J Pathol. 2015; 235:539-545.
- Lee JC, Su SY, Changou CA, Yang RS, Tsai KS, Collins MT, Orwoll ES, Lin CY, Chen SH, Shih SR, Lee CH, Oda Y, Billings SD, Li CF, Nielsen GP, Konishi E, Petersson F, Carpenter TO, Sittampalam K, Huang HY, Folpe AL. Characterization of FN1-FGFR1 and novel FN1-FGF1 fusion genes in a large series of phosphaturic mesenchymal tumors. Mod Pathol. 2016; 29:1335-1346.
- Jan de Beur SM, Miller PD, Weber TJ, Peacock M, Insogna K, Kumar R, Rauch F, Luca D, Cimms T, Roberts MS, San Martin J, Carpenter TO. Burosumab for the Treatment of Tumor-Induced Osteomalacia. J Bone Miner Res. 2021;36(4):627-635.
- de Castro LF, Ovejero D, Boyce AM. Eur J Endocrinol. DIAGNOSIS OF ENDOCRINE DISEASE: Mosaic disorders of FGF23 excess: Fibrous dysplasia/McCune-Albright syndrome and cutaneous skeletal hypophosphatemia syndrome. 2020 May;182(5):R83-R99. doi: 10.1530/EJE-19-0969
- Lim YH, Ovejero D, Sugarman JS, Deklotz CM, Maruri A, Eichenfield LF, Kelley PK, Jüppner H, Gottschalk M, Tifft CJ, Gafni RI, Boyce AM, Cowen EW, Bhattacharyya N, Guthrie LC, Gahl WA, Golas G, Loring EC, Overton JD, Mane SM, Lifton RP, Levy ML, Collins MT, Choate KA. Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Hum Mol Genet. 2014; 23:397-407.
- White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, McKeown C, Fitzpatrick D, Yu K, Ornitz DM, Econs MJ. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005; 76(2):361-367.
- Brown WW, Jüppner H, Langman CB, Price H, Farrow EG, White KE, McCormick KLHypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen’s metaphyseal chondrodysplasia. J Clin Endocrinol Metab. 2009; 94(1):17-20.
- Fradet A, Fitzgerald J. INPPL1 gene mutations in opsismodysplasia. J Hum Genet. 2017;62(2):135-140.
- Tieder M, Modai D, Samuel R, et al. Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med. 1985; 312:611-617.
- Tieder M, Modai D, Shaked U, et al. Idiopathic hypercalciuria and hereditary hypophosphatemic rickets. Two phenotypical expressions of a common genetic defect. N Engl J Med. 1987; 316:125-129.
- Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P, Schnabel D, Hochberg Z, Strom TM. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006; 78:193-201.
- Jaureguiberry G, Carpenter TO, Forman S, Jüppner H, Bergwitz C. A novel missense mutation in SLC34A3 that causes HHRH identifies threonine 137 as an important determinant of sodium-phosphate cotransport in NaPi-IIc. Am J Physiol Renal Physiol. 2008; 295: F371-F379.
- Magen D, Berger L, Coady MJ, Ilivitzki A, Militianu D, Tieder M, Selig S, Lapointe JY, Zelikovic I, Skorecki K: A loss-of-function mutation in NaPi-IIa and renal fanconi's syndrome. N Engl J Med. 2010; 362:1102-1109.
- Schlingmann KP, Ruminska J, Kaufmann M, Dursun I, Patti M, Kranz B, Pronicka E, Ciara E, Akcay T, Bulus D, Cornelissen EA, Gawlik A, Sikora P, Patzer L, Galiano M, Boyadzhiev V, Dumic M, Vivante A, Kleta R, Dekel B, Levtchenko E, Bindels RJ, Rust S, Forster IC, Hernando N, Jones G, Wagner CA, Konrad M. Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol. 2016; 27(2):604-14.
- Scheinman SJ, Pook MA, Wooding C, Pang JT, Frymoyer PA, Thakker RV. Mapping the gene causing X-linked recessive nephrolithiasis to Xp11.22 by linkage studies. J Clin Invest. 1997; 91:2351-2357.
- Scheinman SJ. X-linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney Int. 1998; 53:3-17.
- Gonzalez Ballesteros LF, Ma NS, Gordon RJ, Ward L, Backeljauw P, Wasserman H, Weber DR, DiMeglio LA, Gagne J, Stein R, Cody D, Simmons K, Zimakas P, Topor LS, Agrawal S, Calabria A, Tebben P, Faircloth R, Imel EA, Casey L, Carpenter TO. Unexpected widespread hypophosphatemia and bone disease associated with elemental formula use in infants and children. Bone. 2017; 97, 287-292.
- Eswarakumar AS, Ma NS, Ward LM, Backeljauw P, Wasserman H, Weber DR, DiMeglio LA, Imel EA, Gagne J, Cody D, Zimakas P, Topor LS, Agrawal S, Calabria A, Tebben P, Faircloth RS, Gordon R, Casey L, Carpenter TO. Long-Term Follow-up of Hypophosphatemic Bone Disease Associated With Elemental Formula Use: Sustained Correction of Bone Disease After Formula Change or Phosphate Supplementation. Clin Pediatr (Phila). 2020;59(12):1080-1085.
- Harvey BM, Eussen SRBM, Harthoorn LF, Burks AW. Mineral Intake and Status of Cow's Milk Allergic Infants Consuming an Amino Acid-based Formula. J Pediatr Gastroenterol Nutr. 2017; 65(3):346-349.