ABSTRACT
Viruses are one of the simplest pathogenic organisms infecting the human body. Association between viral infections and endocrine system is complex and has not been fully studied. Viral infections can induce several physiological changes in the human endocrine system, resulting in cytokine mediated activation of hypothalamo-pituitary-adrenal axis to increase cortisol production, thus modulating the immune response. Further, many viral infections impact different endocrine organs, either by direct viral invasion or by systemic or local inflammation resulting in transient or permanent endocrinopathies; both hyper and hypofunction of endocrine organs may ensue. Viruses can encode production of specific viral proteins that have structural and functional homology to human hormones. Since endocrine hormones have immunoregulatory functions, endocrinopathies may alter the susceptibility of human body for viral infections. Recently the pandemic causing SARS 2 CoV infection has been shown to affect multiple endocrine organs through a variety of mechanisms, highlighting the significance of viral infection related endocrinopathies in morbidity and mortality. With improving understanding of viruses and their role in the human endocrine system, further research on this field would be required to explore new targets for prevention and treatment of endocrinopathies.
INTRODUCTION
The endocrine system plays a vital role in homeostasis and immunity. Hormones modulate host defenses by strengthening or weakening the body’s immune system, being one of the key determinants of susceptibility of humans for infections. On the other hand, infective agents through different mechanisms may affect endocrine organs, causing endocrinopathies.
Viruses are the most abundant form of life on earth, estimate at 10 quintillion (1031) (1, 2). They are obligate parasites with single or double-stranded DNA or RNA, infecting a variety of hosts; bacteria, algae, fungi, plants, insects, and vertebrates. Viral infections are common among humans, resulting in diseases which may extend into epidemics. The literature is evolving on viral agents being important pathogens in endocrine disorders. Knowledge about the interplay between viruses and the human endocrine system would expand the understanding of acute and chronic effects of viral disease on the human body.
HUMANS VIROME
The role of viruses in the human body is diverse. Many viruses have been identified to play an essential part in the human microbiome, with an estimate of 380 trillion viral particles inhabiting the human body. This human virome consists of:
- Human Endogenous Retroviruses (HERV): These viruses are integrated viral genomic material which consists of 8% of the total human genome (1, 3). As they are incorporated into the human genome, they are transmitted vertically to offspring. The function of the HERV in the human genome is not known. However, transcription of these genes may produce proteins, which may alter the function of regulatory genes and elicit an immune response (autoimmune diseases) or cell growth (cancers) in the host (4, 5).
- Bacteriophages: These viruses inhabit their bacterial hosts, mainly in the human gut in addition to the skin and oral cavity (6, 7). Mostly they show a symbiotic relationship with their host. They may promote health or disease by phage-mediated lysis of pathogenic or commensal bacteria triggered by external factors, thereby controlling bacterial populations and promoting evolutionary advantages to host bacteria (e.g., antibiotic resistance through the transfer of genetic material)
- Eukaryotic viruses: These viruses infect human cells often resulting in symptomatic diseases or may exist in an asymptomatic carrier state (8).
PATHOGENESIS OF VIRAL INFECTIONS IN HUMANS
Being an obligate parasite, viruses need to enter the human cell in order to replicate and complete its life cycle. The first step of viral entry into a host cell involves recognition and binding to host cell specific receptors by viral surface proteins (fusion proteins/ spikes). This specificity determines the host range for a particular virus (e.g. HIV viral surface protein, gp120 specifically interact only with CD4, CCR5, and CXCR4 molecules on T lymphocytes) (9). Subsequent events are conformational change of fusion proteins and subsequent entry into the cell through endocytosis (e.g. Adenovirus), cell membrane fusion (e.g. HIV) or direct cell to cell contact (virological synapses e.g. HTLV1) (10). Once the viral DNA or RNA material is inside the host cell, they are incorporated into the host genome and replication of the viral genome and/or transcription of specific proteins will occur. The replicated viral genome gets assembled with viral proteins to produce an increased number of viral particles (virions), which will be released from the infected cell by cell lysis or budding from the host cell. Some viruses undergo a lysogenic cycle where the viral genome is incorporated into a specific location in the host chromosome. This viral genome is known as provirus and mostly remain non-functional within host cells until being activated at some point when the provirus gives rise to active virus which would lead to the cellular death, necrosis, or apoptosis of the host cell (9).
Many changes occur in the infected cell and its surrounding tissues leading to immune modulations. Direct cellular damage could occur in the infected host cell by viral proliferation/replication. Proteins encoded by the viral genome can also modulate cellular functions and cause damage to the infected cell (11). Further, the virus itself and infected tissues trigger the innate immune system through cytokines (IL-1, IL-6, IL-8, IL-12, TNF-α, IFN-α/β, TGF-β), complements, and natural killer cells (12, 13). The resultant immune response can cause inflammation or subsequent damage to the infected cell which may further progress to inflammation and damage of nearby uninfected cells (11, 14). Virus induced tissue inflammation increases the production of systemic mediators of inflammation, which may induce a systemic inflammatory condition in the host, affecting many other organs ((11, 15). Subsequently adaptive immunity develops where the host develops cell mediated immunity (to control disease) and humoral immunity with antibodies (to prevent reinfection) against specific viruses (12).
VIRUS INDUCED ALTERATIONS IN THE ENDOCRINE SYSTEM
Virus induced changes of endocrine cells and organs can occur in several ways (11).
- Activation of the hypothalamo-pituitary-adrenal (HPA) axis indirectly as a result of systemic viral infection and inflammation.
- Damage to specific endocrine cells by direct viral infection of the cell (through stages of the viral cell cycle).
- Damage to specific endocrine cells by viral proteins produced within the cell as a result of viral replication within the cell.
- Damage of virus infected endocrine organs by inflammation through activation of an immune reaction (innate and cell mediated).
- Damage of uninfected endocrine organs through the systemic immune response as a result of the immune reaction to the viral infection.
- Damage of uninfected endocrine organs through autoimmune mechanisms/cross reaction of antibodies.
- Viral gene products may induce an alteration of hormonal activity/ production by endocrine cells.
Pituitary Gland
HPA AXIS RESPONSE TO VIRAL INFECTIONS
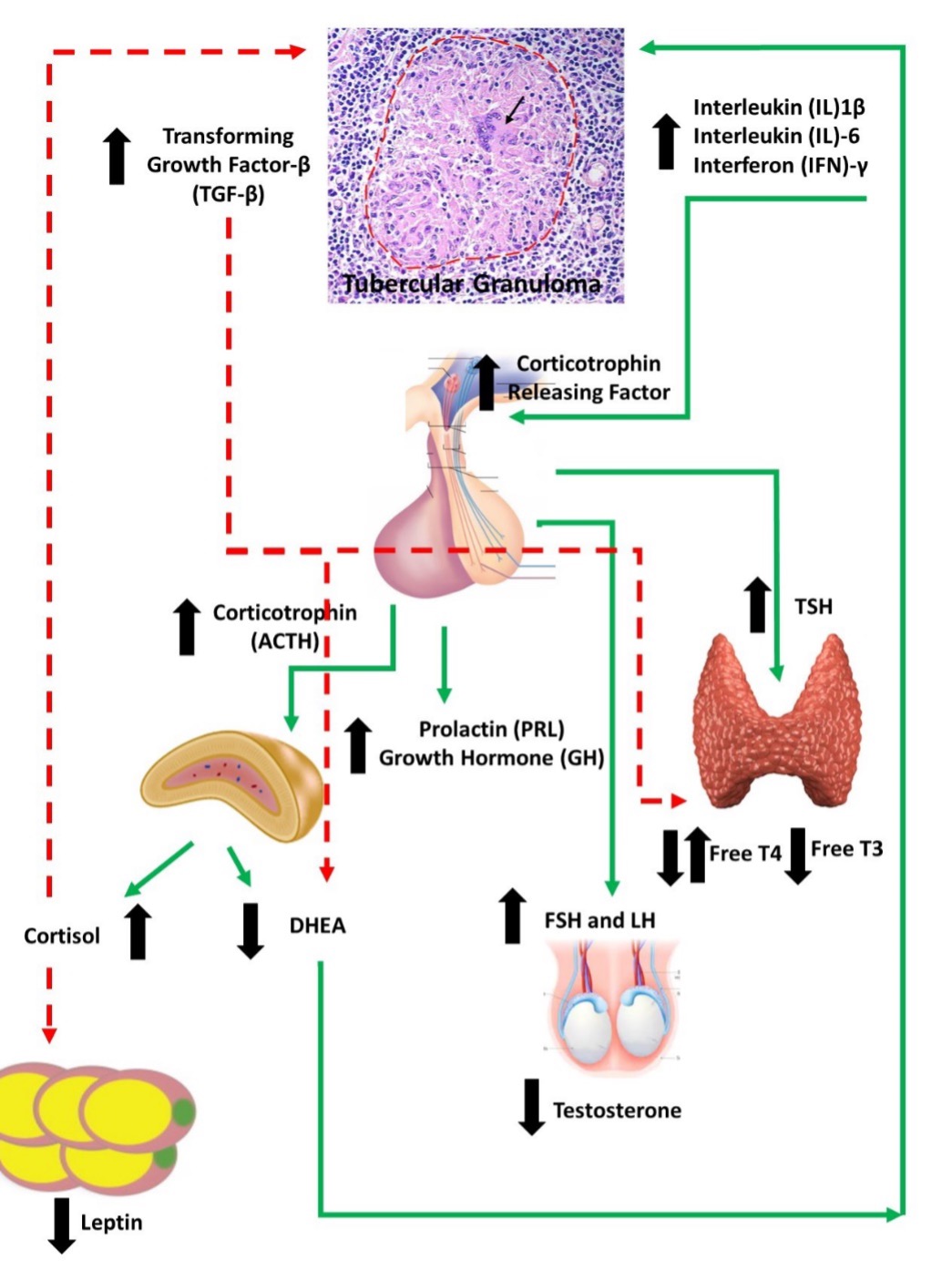
Any virus which causes systemic viremia and inflammation may stimulate the HPA axis to release cortisol through early innate proinflammatory cytokines (IL-1, IL-6 and TNF-α) and late acquired T cell cytokines (INF-ϒ and IL-2) during an acute viral infection (16). These mediators act on the CRH producing cells of the hypothalamus, corticotrophs in the anterior pituitary as well as on the adrenal cortex to increase glucocorticoids during the illness. Cytokines (IL-1, -2, -4, -7, -8, TNFα and INF-α) are responsible for the expression of glucocorticoid receptors on lymphocytes and neutrophils, enhancing the immune reaction (17). While the stress hormones norepinephrine and epinephrine mobilize immune cells into the blood stream, epinephrine and cortisol are responsible for enhancing differentiation of specific immune cells and directing the cells to the tissues where they are needed (18-20). On the other hand, elevations in glucocorticoids negatively regulate the immune system to reduce the further production of cytokines (e.g., HSV-1, MCMV, influenza,) and promote switching of cellular immunity to humoral immunity, thus protecting the body from an overactive immune response (16). The extent of viral induced glucocorticoid production is specific to the virus and the immune response stimulated by the virus; thus, in infections like LCMV clone E350 where no significant inflammation occurs, glucocorticoid production is minimal (21). However, mouse models with corneal inoculation of HSV-1 virus have shown that even without significant viremia, brainstem infection of HSV-1 can alter the HPA axis stress response through central mediated pathways (22).
ANTERIOR PITUITARY DYSFUNCTION
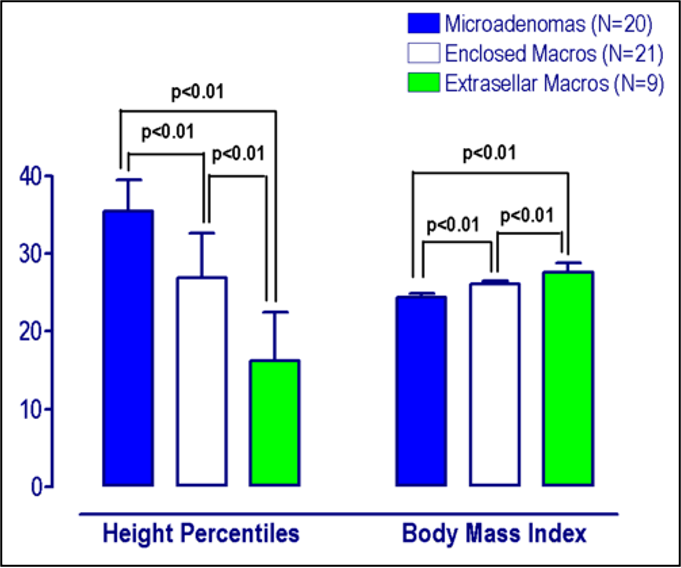
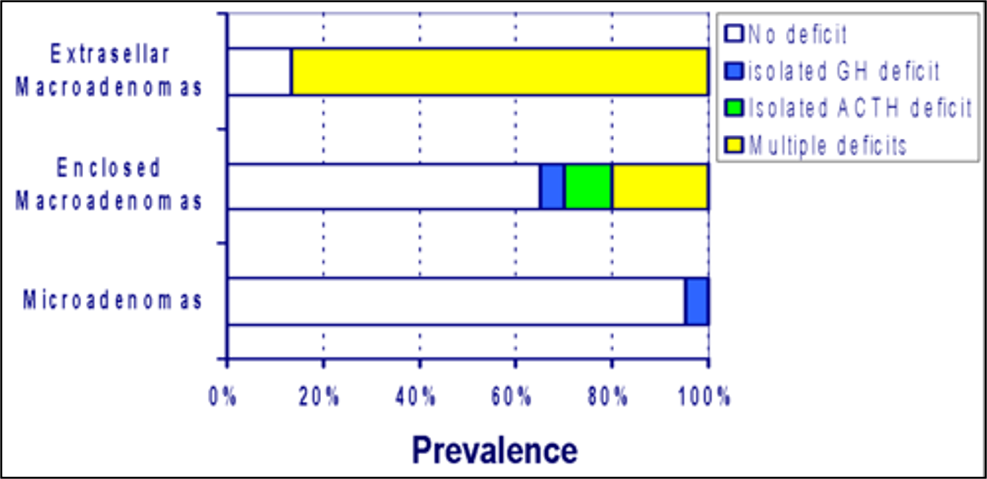
Hypothalamo-pituitary dysfunction due to infections remain underestimated (23). Isolated or multiple anterior pituitary hormone deficiencies had been identified during acute viral illnesses of the central nervous system, especially following viral meningo-encephalitis (24, 25). Pituitary dysfunction mainly involves corticotrophin deficiency and hyperprolactinemia and to a lesser extent gonadotrophin, growth hormone, or thyrotrophin deficiencies (24-27). The prevalence of individual deficiencies following viral infections vary among studies. Patterns of hormonal deficiencies following CNS infections depend on the type of causative agent, the localization of the infection, as well as with the severity of the disease (24). Partial or complete deficiency of specific hormones may occur depending on the degree of damage (28).
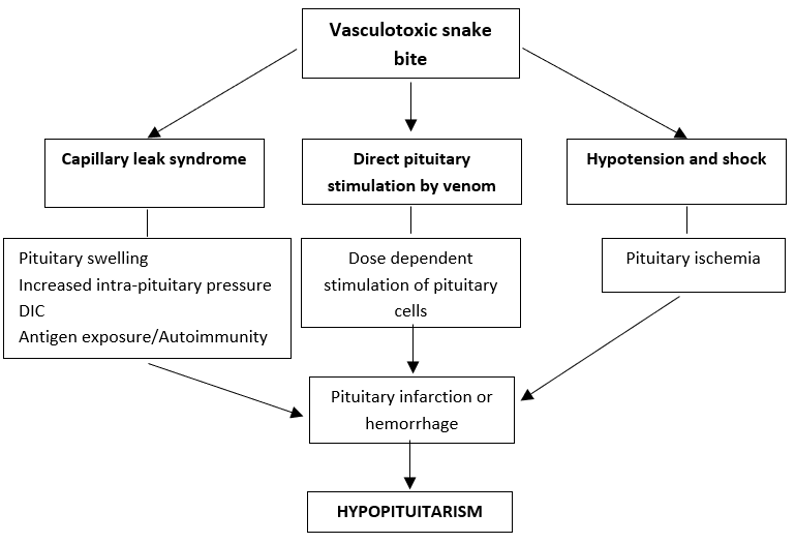
The main mechanisms involved in hypopituitarism with examples are shown in Table 1.
|
Table 1. Virus Induced Changes in Pituitary Function
|
||||
|
Outcome |
Mechanism |
Hormone |
Examples |
Ref |
|
HPA activation |
Cytokines T cell mediated |
Cortisol |
Any virus causing significant inflammation |
(11, 16) |
|
Hypopituitarism |
Hypothalamic involvement |
CRH, GHRH, TRH, GnRH |
CMV, VZV, HIV, SARS |
(29-32) |
|
Hypophysitis by direct viral infiltration |
ACTH, TSH, GH, LH, FSH |
HSV-1/2, HIV, Hanta, SARS |
(29, 32-36) |
|
|
Ischemia/infarction causing apoplexy |
ACTH, TSH, GH, LH, FSH |
Hanta, Influenza A |
(33, 37) |
|
|
Hemorrhage to pituitary (Later empty Sella) |
GH, ACTH, TSH, LH, FSH, |
Hanta, VZV |
(28, 38-40) |
|
|
Antibody mediated hypophysitis |
ACTH, GH, FSH, LH |
SARS-COVID 19, Rubella |
(41, 42) |
|
|
Mechanism not identified |
|
Coxsackie B5, Influenza A, |
(24, 43, 44) |
|
|
Hyperprolactinemia |
Dopaminergic stress response |
Prolactin |
|
(52, 53) |
|
SIADH |
Damage to hypothalamic/ stalk / posterior pituitary cells |
ADH |
HSV-1, EBV |
(45-47) |
|
Diabetes insipidus |
Direct viral invasion |
ADH |
Hanta, CMV, HSV, Coxackie B1 |
(48-51) |
Hypophysitis can occur either early in the acute phase of the infection or at a later stage as a sequela of a CNS infection (24, 25). A proportion of patients who have deficiencies in anterior pituitary hormones during the acute phase of a viral infection recover hormonal function, typically within 1 year, indicating the transient nature of the damage in a subset of patients (25, 29). Delayed development of pituitary insufficiency can remain asymptomatic or present with vague symptoms. The deficiencies in such case will remain permanent, needing life-long hormonal replacement. If not for a high degree of suspicion, hypopituitarism can be misdiagnosed as the post-encephalic syndrome (23).
Acute viral infections can cause hyperprolactinemia in response to elevated cytokines IL-1, IL-2 and IL-6, which stimulate prolactin production (52). Further, certain viral infections (e.g., HIV) directly reduce the dopaminergic tone increasing prolactin levels (53). Macrophages, monocytes, lymphocytes, and natural killer cells possess prolactin receptors. Prolactin binding to these receptors activates downstream signaling pathways that will stimulate immune cell proliferation, differentiation, and survival (52). Prolactin also antagonizes the immunosuppressive effects of TNF-α and TGF-β (54). Therefore, elevated prolactin acts as an immunomodulator during acute infections.
Although rare, CNS viral infections may also present as an emergency with pituitary apoplexy, precipitating ischemia or bleeding in a normal pituitary gland or a pituitary tumor (33, 37, 38, 40). Some patients present with the empty sella syndrome following initial hemorrhagic insult to the pituitary, mostly with anterior pituitary hormonal deficiencies.
POSTERIOR PITUITARY DYSFUNCTION
SIADH is a well-known complication of infections, particularly CNS viral infections with HSV and tick-born infections(55, 56). The possible mechanism is cytokines released during inflammation, especially IL-6, causing non-osmotic release of vasopressin from the posterior pituitary (57). The presence and severity of hyponatremia depends on the infective agent, severity of the infection, and infected foci. Hyponatremia portends a worse prognosis (45).
Direct viral destruction of ADH and oxytocin producing neurons in the hypothalamus, pituitary stork, or posterior pituitary would result in central diabetes insipidus (49). It is usually associated with anterior pituitary hormone deficiency and is described mainly in immunocompromised patients with encephalitis (49, 58, 59).
Thyroid Gland
Systemic viral infections may result in alterations of thyroid functions, giving rise to the “Sick Euthyroid syndrome”, “non-thyroidal illness syndrome”, or “Low T3 syndrome”(60). This is characterized by decreased levels of serum T3 and sometimes thyroxine (T4), without an increased secretion of TSH. Mechanisms involved are elevated circulating cytokines, cortisol, and free fatty acids influencing deactivation of deiodinase-1 enzyme (reduces T4 to T3 conversion), activation of deiodinase-3 enzyme (increased conversion of T3 to rT3), down regulation of hypothalamo-pituitary-thyroid axis resulting in normal or low TSH levels despite low T3 levels, and changes in thyroid binding proteins (61).
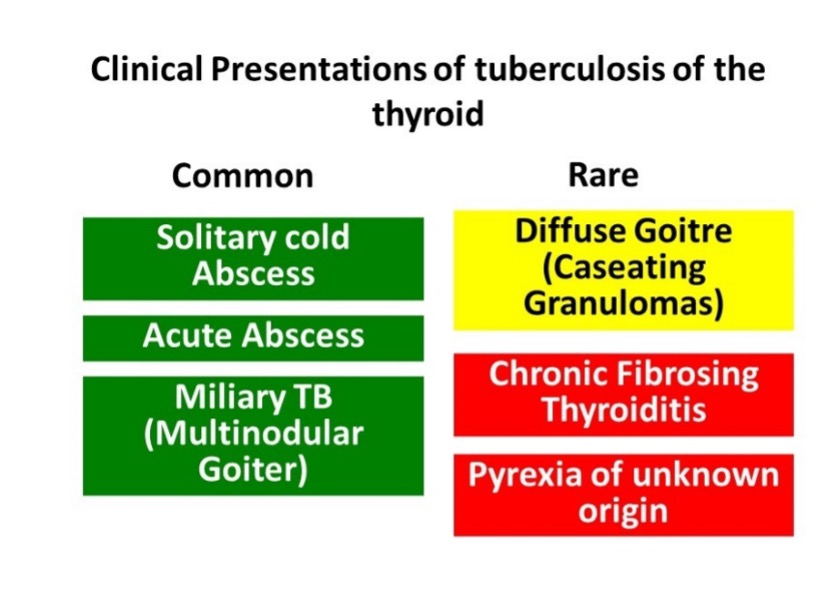
Direct viral invasion of the thyroid gland with subsequent cytotoxic T-cell mediated inflammation of the gland results in infiltration of thyroid follicles with disruption of the basement membrane leading to subacute thyroiditis and the release of thyroid hormones (62). Presence of viral material had been identified in diseased thyroid glands in certain patients (e.g., mumps) while others show viral IgG antibodies / rise in titers (Mumps, Coxsackie, adeno and influenza) (63, 64). However, elevated antibodies to common respiratory tract viruses may be an anamnestic response due to the inflammatory process (65). Epidemiological data has shown subacute thyroiditis during outbreaks of viral infections and seasonal clustering when viral infections are also commonly seen (66).
Viruses triggering the immune response leading to the development of autoimmune thyroid diseases like Hashimoto’s thyroiditis and Grave’s disease has been suggested (67, 68). Possible mechanism include (69);
- Molecular mimicry: recognizing an epitope on an external antigen
- TLR activation by virus and heat-shock protein effects
- Enhanced thyroid expression of human leukocyte antigen molecules
The above mechanisms could facilitate the development of antigen-specific adaptive immunity, causing autoimmune thyroiditis (68, 70). In Grave’s disease, significant homology had been identified in HIV-1 induced viral Nef-protein and the human TSH receptor, raising the possibility of T cell activation through a viral antigen acting as an autoantigen (71). Further, EBV infection results in over-expression of HLA antigens, predisposing to immune mediated mechanisms of Grave’s disease (72).
Elevated levels of TBG had been observed in patients with HIV in both stable and ill patients (36). The mechanism of this finding is unknown but care should be taken in interpreting total T4/T3 levels in patients with HIV.
|
Table 2. Virus Induced Diseases of the Thyroid Gland |
||||
|
Outcome |
Mechanism |
Hormone |
Example |
Ref |
|
Sick euthyroid syndrome |
Deactivation of deiodiase-1 (through cytokine mediated inflammation) |
Low T3 Normal or low T4/TSH |
Any virus causing significant inflammation |
(32, 61) |
|
Activation of deiodinase-3 |
||||
|
Down regulation of HPT axis |
||||
|
Changes in circulating proteins |
||||
|
Subacute thyroiditis |
Direct viral invasion |
Hyperthyroidism followed by hypothyroidism (mostly reversible within 12 months) |
Mumps, Coxsackie, adeno, influenza, SARS |
(63, 64, 73) |
|
Autoimmune hypothyroidism |
Development of adaptive immune system through: - Molecular mimicry - TLR activation - HLA over expression
Antibody mediated |
Overt hypothyroidism (Low T4/T3, Raised TSH |
Parvovirus B19, mumps, rubella, coxsackie, HSV, Hep C, E, EBV |
(66-68) |
|
Subclinical hypothyroidism (normal T4/T3, raised TSH) |
||||
|
Autoimmune hyperthyroidism (Grave’s Disease) |
Hyperthyroidism (Low TSH, raised T4/T3) |
EBV, HIV-1 |
(71, 72, 74) |
|
Parathyroid Glands and Calcium Metabolism
Although rare, parathyroid dysfunction following viral infections has been documented in case reports. Acute hyperparathyroidism has been reported with acute Hepatitis B infection, where hypercalcemia resolved following resolution of the hepatitis. It was postulated that antibody mediated lowering of the calcium set point in the parathyroid gland had resulted in hyperparathyroidism (75).
Hypoparathyroidism is documented with several viral infections including HIV and SARS-COVID 19 (76-78). Parathyroid cells express a protein recognized by antibodies against CD4, the HIV-1 receptor. Therefore, it is possible for the virus to directly infect the parathyroid cells and also circulating autoantibodies against CD4, may impaired PTH release though direct interaction (76).
Hypocalcemia is a well-recognized metabolic derangement in some viral infections (e.g., Dengue, measles, SARS-COVID-19), indicating severe disease and worse outcomes (79-81). This is usually transient during the active disease and could be secondary to vitamin D deficiency, hypoalbuminemia secondary to infection, calcium influx into damaged cells, or hypoxia induced cytokine release mediating impaired PTH secretion or tissue response to PTH (82).
Pancreas
TYPE 1 DIABETES
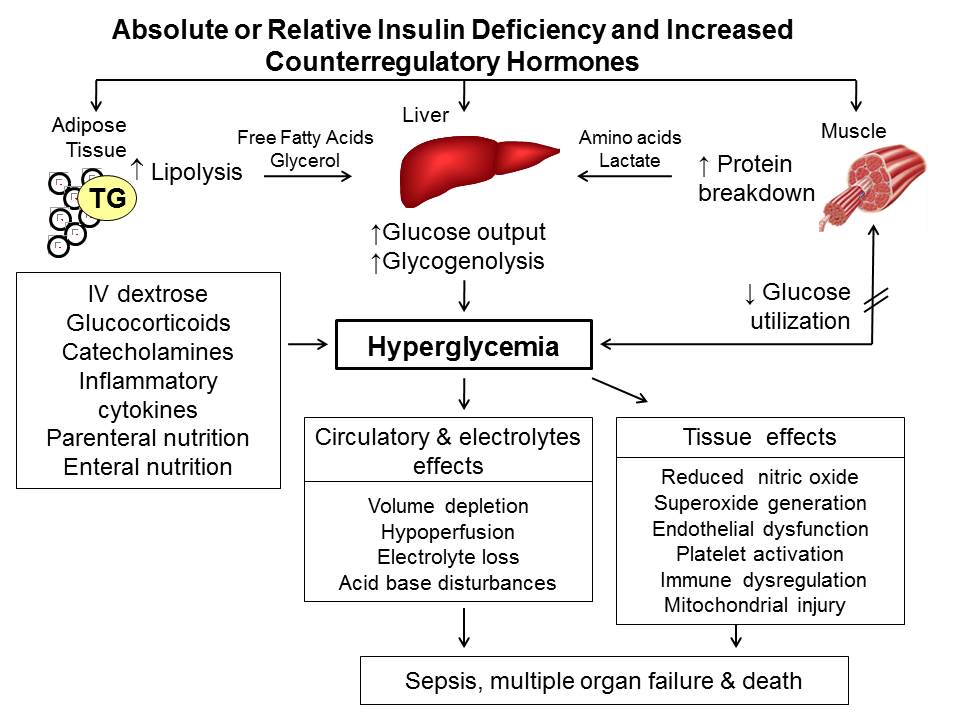
Different viral infections are linked as a potential trigger for the development of Type 1 diabetes. This happens through different mechanisms; direct pancreatic β-cell lysis or immune mediated progressive β-cell destruction though autoimmunity generated from molecular mimicry, bystander activation of autoreactive T cells, and loss of regulatory T cells (83). As a result, hyperglycemia / new-onset insulin dependent diabetes develops.
Direct β-cell damage can occur as a result of viral invasion resulting in beta-cell lysis (e.g. coxsackieviruses, Rubella, mumps, enterovirus, influenza, Hep C (84-87) or inflammation following pancreatitis secondary to viral infection (hepatotropic virus, coxsackie virus, CMV, HSV, mumps, varicella-zoster virus), systemic inflammation, or immunomodulation (88). Although the incidence of diabetes following acute pancreatitis is as high as 23% (89), the incidence following viral pancreatitis had not been studied.
Virus induced autoimmunity appears to be the main mechanism for viral induced type 1 diabetes. However, the role of viruses seems to be more complex. Certain virus infections induce upregulation of MHC class I on β cells, thereby enhancing recognition of β cells by autoreactive CD8+ cytotoxic T lymphocytes, thus inducing autoimmunity (90). However, some virus strains (LCMV, CVB) reduce the incidence or delay the onset of type 1 diabetes, probably through a TLR mediated mechanism (90). Whether inductive or protective autoimmunity depends on several factors; level of infection (more severe infections enhance diabetes while low-replicating strains of the same infection seem protective), genetic predisposition, presence of other detrimental environmental triggering factors like viruses, and direct β-cell toxins (91). Adding to the complexity, research on the intestinal virome has shown that certain changes in the intestinal virome precede autoimmunity, especially in developing type 1 diabetes in genetically susceptible people (92).
To-date, literature support viral infection (e.g. enterovirus, influenza virus, cytomegalovirus, mumps, rubella, rotavirus, and coxsackie virus) mediated autoimmune destruction of beta-cells through molecular mimicry or activating cross-reactive T cells as one of the potential mechanisms for the pathogenesis of type 1 diabetes (93).
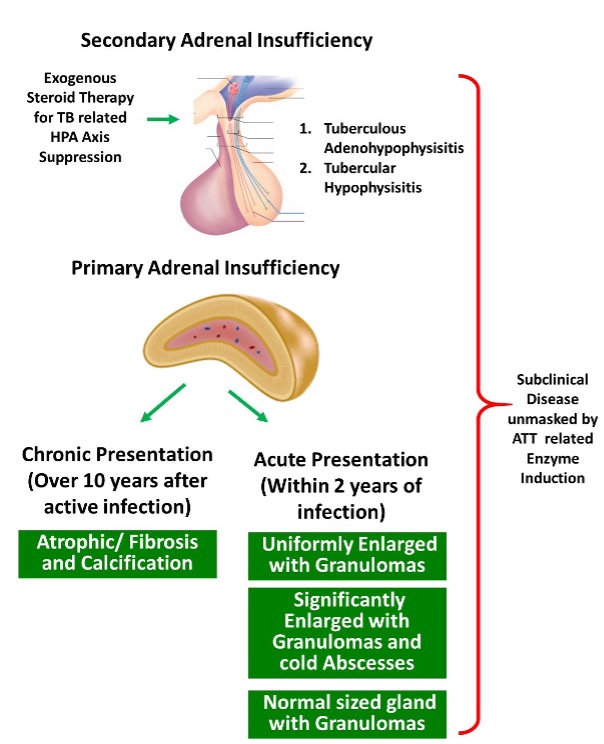
Adrenal Glands
Primary adrenal insufficiency is a well-known consequence of certain viral infections. Common pathological mechanisms are direct viral invasion causing adrenalitis, adrenal hypofunction due to immunological impact from systemic inflammatory response, and viral-induced autoimmune destruction of the adrenal gland (94). Although less frequent, adrenal glands can be infected in early HSV-1 and -2 infections irrespective of the immune status of the host, with the adrenal gland having the highest number of viral particles of any organ (95). SARS-COVID 19 virus also affects the adrenals by direct cytopathic effects by the virus or due to the systemic inflammatory response (96). Antibody mediated adrenal insufficiency is also seen in SARS viral infection by producing molecular mimics to ACTH. Antibodies to the viral peptides bind both viral protein and host ACTH, may destroy ACTH or reduce the functionality of the hormone, thus reducing the ability of ACTH to induce cortisol production (97).
More severe forms of adrenal disruption (usually bilateral) are seen in less common fulminant viral infections; H5N1 avian influenza causing multifocal necrosis of adrenal cells (98), filoviruses (e.g. Ebola) giving rise to liquefaction of the adrenals (99). Further, immunosuppression has been identified as a predisposing factor for severe adrenal infections. Echoviruses serotypes 6 and 11 which cause lethal disseminated intravascular coagulation in children can affect the adrenals with hemorrhagic necrosis (100). CMV adrenalitis had been reported in more than half of the patients with AIDS, with or without evidence of CMV viremia (101, 102).
Adrenal glands are the most commonly affected endocrine organ by HIV infection (103, 104). Early stages of HIV result in a rise in cortisol secretion, as an adaptive response to a stressor. In some patients, this rise in cortisol levels may precipitate reactivation of EBV. During the latter stages, adrenal ‘burnout’, direct viral infection, co-infection by opportunistic microbes (viral – CMV, bacterial, fungal), anti-adrenal cell antibodies (unique to HIV infection), and increased peripheral cortisol resistance may lead to progression to overt adrenal failure (105-107). An autopsy series has shown the adrenal gland to be pathologically compromised in 99.2% of cases of patients with AIDS, highlighting the degree of damage in immunocompromised state (108).
Some adrenal neoplasms have a viral etiological relationship, particularly in HIV infected people; EBV associated lymphoma of the adrenal gland and AIDS-associated neoplasms (e.g. Kaposi sarcoma from HHV-8, non-Hodgkin's lymphoma) (109).
Gonads
TESTES
Viruses are well known to cause orchitis, both unilateral and bilateral, as a consequence of a systemic viral infection. Mumps virus is the commonest viral infection affecting the testes, being the most common complication of mumps in post-pubertal men (110). Virus attacks the testicular tissues, leading to an innate immune response, inflammation, and perivascular intestinal lymphocyte infiltration. Resultant swelling exert pressure on intratesticular tissues and leads to testicular atrophy (111, 112). In addition to having varying degrees of hypogonadism, viral infection leads to subfertility; transient changes in sperm count, mobility, and morphology of fertility in unilateral disease while 30 – 87% having infertility due to oligo-asthenospermia in bilateral disease (113). Other viruses have also been identified to cause direct testicular damage including coxsackievirus, varicella, echovirus, Hep E, Zika virus, and cytomegalovirus, leading to varying degree of testicular damage but to a lesser extent than mumps virus (114-116). Certain viruses have also been detected in semen (e.g., Zika, Ebola, HIV, Hep B, Herpes and SARS-COV) and within spermatozoa (e.g., HIV, Hep B and Zika) of infected patients (117, 118). Although some of these could contribute to vertical transmission as well as affect fertility, the role of the viruses needs to be clarified with further research (119, 120).
HIV infects the testis early during the course of the disease, targeting testicular leucocytes and germ cells, but is not associated with any apparent morphological changes (121). Viral infection of the germ cells is important in vertical transmission of the disease. Further, HIV-2 and SIV but not HIV-1 are known to damage Leydig cells and reduce testosterone levels (122). HIV/AIDS also make the testes more susceptible for opportunistic infections like CMV, EBV and TB (123). Some evidence has emerged for an oncogenic effect of HIV and EBV infection on human testis but further research is needed (124).
Although viral orchitis induces humoral immune response and result in anti-sperm antibodies, their causal link to subfertility or hypogonadism is unclear (110, 125).
OVARIES
Viruses affect ovaries through similar mechanisms as the testes; direct invasion and innate immune mediated oophoritis, resulting in changes in estrogen/progesterone to cause varying degrees of hypogonadism as well as affect fertility (126, 127). Mumps, Zika, HIV, and CMV viruses have been documented in the literature as common viral culprits affecting the ovaries and the clinical manifestations range from being silent infections to oophoritis resulting in premature menopause (126-128).
Further, some viruses (e.g. CMV) had been implicated in ovarian carcinogenesis but further studies need to clarify the exact causal effect and pathogenesis (129).
Viral Protein Mediated Modulation of Hormone System
New insights have shown that the viral genomes can produce viral peptide sequences that possess homology with human hormones, growth factors, and cytokines. Such viruses may affect human physiology not by infecting and damaging tissues nor by eliciting immune responses but by producing molecules which mimic the action of functional molecules. Recent research pointed out that viruses belonging to Iridoviridae family, which commonly infect fish, but have been also identified in the human virome (blood and feces), produce viral insulin like peptide (VILP), which has homology to human insulin/ IGF-1 (130). These VILPs could compete with endogenous ligands, stimulate or impair post receptor signaling in an autocrine, paracrine, and endocrine basis. They bind to human receptors and stimulate downstream signaling, increase glucose uptake by adipocytes in vitro and in vivo, and stimulate cell proliferation and growth, but less potently than human insulin/IGF-1 (130-132). The role of VILPs are not fully understood and further research is needed to explore the links between VILP and T1DM, insulin resistance, and certain neoplastic growths in association with viral infections.
Other “viral hormones” have also been identified, with structural and/or functional homology to human hormones including IGF-1 and IGF-2, endothelin-1 (ET1), endothelin-2, TGF-β1 and TGF-β2, fibroblast growth factors 19 and 21, inhibin, adiponectin, resistin, adipsin, and irisin in various viruses (132). However, their exact role in altering human physiology in a beneficial or harmful manner is yet to be identified.
Certain virus genome encoded proteins may alter the human genomic expression of particular hormones. A classic example is HTLV-1 infected cells producing PTHrP causing hypercalcemia by trans-activation of the PTHrP gene through the TAX viral gene product (133). Certain viral products act at the cellular receptor/post receptor level, resulting in alterations of intracellular hormonal signaling pathways. HIV-1 related Vpr and Tat protein may induce hypersensitivity of the glucocorticoid receptor (GR) resulting in lipodystrophy and insulin resistance (134). A similar mechanism is suggested for Paget’s disease, where there is a possible mumps viral peptide (Nucleocapsid transcript) induced hypersensitivity of osteoclastic receptors to vitamin D (135). Inactivation of hormone receptor / post receptor signaling may be seen with some viruses; RSV protein miR-29a down regulates GR receptors (136); poxvirus MCV MC013L protein induces inhibition of glucocorticoid nuclear receptor transactivation (137), and E1A protein produced by Adenovirus blocks the action of glucocorticoids on transcription activity genes (138) resulting in resistance to glucocorticoids. As illustrated by the above examples, viral encoded proteins may affect the human endocrine system through alteration of endocrine signaling systems.
VIRAL INFECTIONS AS A COMPLICATION OF ENDOCRINE DISEASES
HPA Axis
Patients with untreated or undertreated adrenal insufficiency are at a higher risk of infections as well as 5-fold higher mortality from infection compared to the normal population (139, 140). Impaired innate immune function, especially with lower natural killer cells among patients with adrenal insufficiency has the potential to make them more susceptible to invading pathogens and cause a higher rate of death following viral infections (141-143). On the other hand, among patients with undiagnosed glucocorticoid deficiency, the initial HPA axis response to increase the level of glucocorticoids in response to the immune mediators will be altered, hence appropriate shaping of the downstream immune response will not take place. This may result in overactive immune response to the viral infections, affecting mortality and morbidity. Replacement regimes which restore physiological glucocorticoid secretion patterns have been shown to reduce susceptibility to infections as well as to improve mortality among patients with adrenal insufficiency (144). Therefore, appropriate steroid treatment regimens should be employed in patients with deficiencies in the HPA axis depending on their clinical state (Doubling steroid dose in mild viral infections, up to 200mg/24-hours hydrocortisone in moderate-severe viral infections) (145).
Stress induced hypercortisolemia and elevated epinephrine levels, which occur as a physiological mechanism, could mediate latent viral reactivation (HSV, EBV, VZV) through IL-6 mediated pathways, precipitating viral infections (146-148). However, the level of stress may also determine the threshold for reactivation (e.g. low stress levels precipitating VZV, high stress levels precipitate an increase in HSV-1 DNA load) (148).
Viral infections have an increased prevalence and more severe disease course among patients who are immunosuppressed compared to the general population (149). Cushing’s syndrome or over treatment with steroids impair immune function and are considered as an immunocompromised state with high susceptibility for severe viral infections. Further, the high glucocorticoid levels may mask the initial symptoms of viral infection, delaying medical treatment and would lead to a clinical course which is more difficult to predict than in the normal population (150, 151). In addition, concordant viral infection may precipitate adrenal crisis in patients on glucocorticoid replacement therapy, which adds to the increased mortality from viral illness (139).
Pituitary
HYPOPITUITARISM
Several anterior pituitary hormones have immunoregulatory roles (152). GH, PRL, TSH, and gonadotrophins show stimulatory effects on immune cells while gonadal steroids tend to suppress immune mechanisms (153-156). Patients with multiple deficiencies of anterior pituitary hormones would therefore have variable alterations in their immune systems and show an increased susceptibility for infections and an increased tendency to develop serious infections (157).
HYPERPROLACTINEMIA
Elevated prolactin levels act as immunomodulators and enhance the host innate and adaptive immune system. Thus, prolactin may protect the host from viral infections as well as help in containing viral infections (52-54).
Thyroid Gland
Thyroidal diseases have not been shown to increase the susceptibility to viral infections.
Parathyroid Glands
HYPERPARATHYROIDISM
Several in vivo and in vitro studies have demonstrated the immunomodulatory effect of parathyroid hormone. PTH receptors had been identified on cells involved in immune pathways, including neutrophils and B and T lymphocytes (158). In patients with CKD and secondary hyperparathyroidism, elevated PTH has been shown to affect immune cells causing impaired migration, reduced phagocytic and bactericidal activity, and inhibited granulocyte chemotaxis (159, 160). T and B lymphocyte proliferation and antibody production are also affected by PTH. Thus, PTH may reduce the host response to viral infections and increase the susceptibility for severe viral infections (159).
Gonads
Gender had been one of the key epidemiological factors determining the prevalence and intensity of human viral infections, with men tending to be more susceptible (161). It was shown that females mount a stronger innate and adaptive immune response than males, giving rise to the above differences (162). Rodent studies had demonstrated that in addition to the influence of the sex chromosomes on immune cells, sex hormones also account for this difference through their effects on the immune systems by influencing cell signaling pathways resulting in differential production of cytokines / chemokines and also enhancing T-cell populations and adaptive immune systems through receptors on immune cells (163-165).
Low testosterone levels negatively impact the outcome of certain viral infections. Low levels seen in elderly men and in young men with hypogonadism result in higher morbidity and a worse clinical course following certain viral infections (e.g. Influenza) and antibody response is reduced compared to young healthy males (166). Furthermore, treatment with testosterone has been shown to reduce mortality and improve disease course in both young and elderly males with hypogonadism (166). These changes were mainly due to alterations in the immune response by testosterone receptor mediated leukocyte contraction to limit inflammation (monocytes, CD8+ T cells, degranulation and reduced cytokine production) rather than limiting viral replication (165, 167).
Estrogen acts as a potent anti-inflammatory hormone that reduces the severity of viral infections such as the influenza virus. Estrogen alters the production of chemokines, target organ recruitment of neutrophils, and cytokine responses of virus-specific CD8 T cells to protect against severe influenza (168). Estrogen shows bipotential effects; small or cyclical amounts enhancing proinflammatory cytokine responses and high or sustained circulating levels reducing production of proinflammatory cytokines and chemokines (169). Female hypogonadism in the form of natural menopause or pathological hypogonadism in young women has been shown to increase susceptibility for viral infections while the use of hormonal replacement therapy or estrogen replacement has reduced viral infections and hospital admissions (170).
Diabetes
Chronic hyperglycemia is known to be associated with altered innate immune response with reduced chemotaxis, phagocytosis, killing by polymorphonuclear cells and monocytes/macrophages, a reduced response of T cells, reduced neutrophil function, and varying disorders of humoral immunity (171). These defects in immune response predisposes patients with diabetes, especially individuals with poor glycemic control, to different viral infections as well as results in a more severe disease (172). Respiratory viruses like influenza are known to affect patients with diabetes commonly and have a sixfold higher risk of hospitalization compared to non-diabetics (173). Further, SARS-CoV-2 virus also tends to result in more severe infections in patients with poorly controlled diabetes (174).
ENDOCRINE PARAMETERS AS MARKERS OF SEVERE VIRAL INFECTION
Severe systemic viral infections tend to cause changes in thyroid profile, which is known as Non-Thyroidal Illness (NTI)/ Sick Euthyroid syndrome. Classic changes in the thyroid profile are low T3 and normal/low TSH and fT4 levels. Low T3 levels and the presence of NTI has been associated with severe viral infections and poor outcomes in several viral infections (e.g., CNS viral infections, SARS-Cov-2) (60, 175).
Among many other markers of disease severity, low serum calcium level is also a predictor for severe disease and worse clinical outcome among patients with Dengue and SARS-COV-2 infections (82, 176, 177). The above markers could be used in addition to the usual clinical parameters for assessment and prediction of disease severity and prognosis.
COVID-19 AND THE ENDOCRINE SYSTEM
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus has spread across the globe rapidly since the end of 2019 (Corona virus disease 2019; COVID-19), causing an unprecedented pandemic with significant mortality and morbidity. Because of the novelty of the disease, the possible impact on the endocrine system is not fully understood. However, emerging evidence support that COVID-19 has significant effects on the endocrine system. Additionally, patients with certain endocrinopathies face a worse outcome from COVID-19 infections.
SARS-CoV-2 virus uses the host angiotensin-converting enzyme 2 (ACE2) as the receptor for fusion and entry into human cells in order to complete its life cycle (178). Lung is the principal target of COVID-19 virus due to an abundancy of ACE2 receptors and is responsible for the main symptoms of the disease but many endocrine organs (hypothalamus, pituitary, thyroid, pancreas, adrenals, gonads) also express ACE2 receptors abundantly and this predisposes viral entry into endocrine organs and subsequent viral induced changes (179, 180). Possible pathogenic mechanisms are direct viral entry and destruction of cells, inflammation induced cellular dysfunction, and immune/antibody mediated hormonal dysfunction.
COVID-19 and Endocrinopathies
Data on the impact of COVID-19 infections on endocrine organs are emerging. Previous SARS viral infection outbreaks have revealed a spectrum of endocrinopathies which span from asymptomatic disease to gross hypofunction of endocrine organs (hypothalamus, pituitary, thyroid, adrenals, pancreas and gonads). Considering the similarities of other SARS viruses and SARS-Cov-2 (COVID-19) in structure and pathogenic mechanisms, we can assume that a similar spectrum of diseases can be expected during COVID-19 pandemic. Table 3 outlines a summary of available data on the impact of COVID-19 infection on endocrine organs.
|
Table 3. COVID-19 Infection Related Endocrinopathies |
|||
|
Gland |
Outcome |
Mechanism |
Reference |
|
Hypothalamus / Pituitary |
Hypopituitarism (Hypocortisolism / Secondary hypothyroidism) |
Hypothalamic involvement |
(29)* (181, 182) |
|
Hypophysitis from direct viral infiltration |
(29)*, (183) |
||
|
Molecular mimicry for ACTH inducing antibody production, with subsequent destruction of ACTH |
(97)* |
||
|
Central diabetes insipidus |
Hypoxic Encephalopathy following COVID-19 pneumonia Autoimmune neuroendocrine derangement |
(184) |
|
|
SIADH |
Through cytokines - Non-osmotic release of ADH - Hypoxic pulmonary vasoconstriction pathway inducing ADH release Intravascular volume depletion inducing non-osmotic baroreceptor activation Pain inducing ADH release through hypothalamus |
(185-187) |
|
|
Thyroid gland |
Primary hypothyroidism |
Direct viral invasion Consequence of subacute thyroiditis |
(29)* (188, 189) |
|
Subacute thyroiditis |
Direct viral invasion and inflammation |
(188, 189) |
|
|
Grave’s disease |
Virus induced trigger for autoimmunity |
(190) |
|
|
Sick euthyroid syndrome |
Cytokine induced dysregulation of deiodinases |
(190, 191) |
|
|
Parathyroid gland |
Data limited |
||
|
Pancreas |
Diabetes † (Type 1 & Type 2) |
Inflammation / cytokine activation and resultant insulin resistance ‡ |
(192)
|
|
Viral invasion and destruction of islet cells Development of autoimmunity against islet cells ‡ |
(193) |
||
|
Pancreatitis |
(194) |
||
|
Diabetes ketoacidosis |
Worsening of pre-existing diabetes by above ‡ mechanisms New onset of type 1 diabetes |
(195) |
|
|
Adrenal glands |
Primary adrenal insufficiency |
Adrenal necrosis and vasculitis from direct cytopathic effect or inflammatory response |
(196) |
|
Bilateral adrenal hemorrhage |
(197) |
||
|
Gonads (Testis) |
Epididymo-orchitis |
Inflammation / direct viral invasion |
(198) |
|
Hypogonadism †
|
Direct testicular damage by the virus Indirect inflammatory/immune response in the testicles |
(199, 200) |
|
|
Impairment of sperm quality † |
Direct inversion of testes/seminiferous tubules / semen Elevated immune response in testis Autoimmune orchitis |
(201) |
|
|
Gonads (Ovary) |
Data limited |
||
* Evidence based on SARS-Cov-1/ SARS virus infections
† Need further scientific evidence to establish the effect of infection on endocrine dysfunction
In addition to the COVID-19 virus induced hormonal changes, certain pre-existing endocrinopathies may influence the disease course of COVID-19. Patients with immunocompromise due to active Cushing’s disease or long term poorly controlled diabetes are more prone for severe COVID-19 infections and have an increased morbidity and mortality (174, 202). Therefore, care must be taken to treat these patients to achieve good control of their disease state as well as to recognize the increased severity of COVID-19 infections. Patients with adrenal insufficiency are also considered to have an increased risk of more severe COVID-19 infections with a higher complication risk, including adrenal crisis and mortality (139, 203). Careful management of glucocorticoid replacement would minimize the risk.
CONCLUSION
Viruses, being one of the common infective agents affecting humans, play an important role related to physiological and pathological changes in the endocrine system. Through different mechanisms; direct and indirect, viruses influence endocrine organs causing hypo- or hyperfunction. Such changes can result in transient or permanent changes in endocrine glands. On the other hand, certain endocrinopathies may affect the course of viral infections, by altering immune mechanisms. Therefore, thorough knowledge of the interaction of viruses with the endocrine system is important and further research is warranted to gain more detailed insights.
REFERENCES
- Microbiology by numbers. Nature Reviews Microbiology. 2011;9(9):628-.
- Rohwer F, Prangishvili, D. Lindell, D. Roles of viruses in the environment. . Environ Microbiol. 2009;11:2771–4.
- D.J. G. Endogenous retroviruses in the human genome sequence. Genome Biology 2001;2:171.
- Nexø BA, Villesen P, Nissen KK, Lindegaard HM, Rossing P, Petersen T, et al. Are human endogenous retroviruses triggers of autoimmune diseases? Unveiling associations of three diseases and viral loci. Immunologic research. 2016;64(1):55-63.
- Gonzalez-Cao M, Iduma P, Karachaliou N, Santarpia M, Blanco J, Rosell R. Human endogenous retroviruses and cancer. Cancer biology & medicine. 2016;13(4):483-8.
- Hannigan GD, Meisel JS, Tyldsley AS, Zheng Q, Hodkinson BP, SanMiguel AJ, et al. The human skin double-stranded DNA virome: topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome. mBio. 2015;6(5):e01578-15.
- Abeles SR, Robles-Sikisaka R, Ly M, Lum AG, Salzman J, Boehm TK, et al. Human oral viruses are personal, persistent and gender-consistent. The ISME journal. 2014;8(9):1753-67.
- Guerrant RL, Walker DH, Weller PF. Tropical Infectious Diseases: Principles, Pathogens and Practice E-Book: Elsevier Health Sciences; 2011.
- N. James MacLachlan EJD. Virus Replication.Fenner's Veterinary Virology. 5th Edition ed: Academic Press; 2017. p. 17-45.
- Dimitrov DS. Virus entry: molecular mechanisms and biomedical applications. Nature Reviews Microbiology. 2004;2:109-22.
- Kino T, Chrousos GP. Virus-mediated modulation of the host endocrine signaling systems: clinical implications. Trends Endocrinol Metab. 2007;18(4):159-66.
- Scott N. Muller BTR. Immune response to viruses. Clinical immunology. 2008:421-31.
- Wittek A.E. QGVJ. Immunology of viral infections In: R.B B, editor. Textbook of Human Virology 2nd edn. 2nd edition ed: Mosby-Year Book; 1991. p. 116–55.
- Vemuri RS, E.M.; Chieppa, M.; Eri, R.; Kavanagh, K. . Beyond Just Bacteria: Functional Biomes in the Gut Ecosystem Including Virome, Mycobiome, Archaeome and Helminths. Microorganisms 2020;8:483.
- M.B. O. Molecular mimicry and immune-mediated diseases. FASEB J. 1998;12:1255–65.
- Silverman MN, Pearce BD, Biron CA, Miller AH. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005;18(1):41-78.
- Leung DY, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, et al. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. The Journal of experimental medicine. 1997;186(9):1567-74.
- Dhabhar FS, McEwen BS. Stress-induced enhancement of antigen-specific cell-mediated immunity. The Journal of Immunology. 1996;156(7):2608-15.
- Dhabhar FS, Malarkey WB, Neri E, McEwen BS. Stress-induced redistribution of immune cells—From barracks to boulevards to battlefields: A tale of three hormones–Curt Richter Award Winner. Psychoneuroendocrinology. 2012;37(9):1345-68.
- Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses cell-mediated immunityin vivo: A potential role for leukocyte trafficking. Brain, behavior, and immunity. 1997;11(4):286-306.
- Miller AH, Spencer RL, Pearce BD, Pisell TL, Tanapat P, Leung JJ, et al. 1996 Curt P. Richter Award. Effects of viral infection on corticosterone secretion and glucocorticoid receptor binding in immune tissues. Psychoneuroendocrinology. 1997;22(6):455-74.
- Ben-Hut T, Conforti N, Itzik A, Weidenfeld J. Effects of HSV-1, a neurotropic virus, on the hypothalamic-pituitary-adrenocortical axis in rats. Brain Research. 1995;702(1):17-22.
- Pekic S, Popovic V. DIAGNOSIS OF ENDOCRINE DISEASE: Expanding the cause of hypopituitarism. European journal of endocrinology. 2017;176(6):R269-r82.
- Schaefer S, Boegershausen N, Meyer S, Ivan D, Schepelmann K, Kann PH. Hypothalamic-pituitary insufficiency following infectious diseases of the central nervous system. European journal of endocrinology. 2008;158(1):3-9.
- Tsiakalos A, Xynos ID, Sipsas NV, Kaltsas G. Pituitary Insufficiency after Infectious Meningitis: A Prospective Study. The Journal of Clinical Endocrinology & Metabolism. 2010;95(7):3277-81.
- Dhanwal D, Kumar S, Vyas A, Saxena A. Hypothalamic pituitary dysfunction in acute nonmycobacterial infections of central nervous system. Indian Journal of Endocrinology and Metabolism. 2011;15(7):233-7.
- Tanriverdi F, Alp E, Demiraslan H, Dokmetas HS, Unluhizarci K, Doganay M, et al. Investigation of pituitary functions in patients with acute meningitis: a pilot study. Journal of endocrinological investigation. 2008;31(6):489-91.
- Partanen T KM, Leisti P, Salmela P, Pääkkö E, Karttunen A, Sintonen H, Risteli L, Hautala N, Vapalahti O, Vaheri A, Kauma H, Hautala T. . Long-term hormonal follow-up after human Puumala hantavirus infection. Clin Endocrinol (Oxf) 2016;84:85-91.
- Leow MK KD, Ng AW, Ong KC, Kaw GJ, Lee LS. Hypocortisolism in survivors of severe acute respiratory syndrome (SARS). Clin Endocrinol (Oxf). 2005;63(2):197-202.
- Sullivan WM, Kelley GG, O'Connor PG, Dickey PS, Kim JH, Robbins R, et al. Hypopituitarism associated with a hypothalamic CMV infection in a patient with AIDS. The American Journal of Medicine. 1992;92(2):221-3.
- Chapter 20 Hypothalamic infections. In: Swaab DF, editor. Handbook of Clinical Neurology. 80: Elsevier; 2004. p. 91-9.
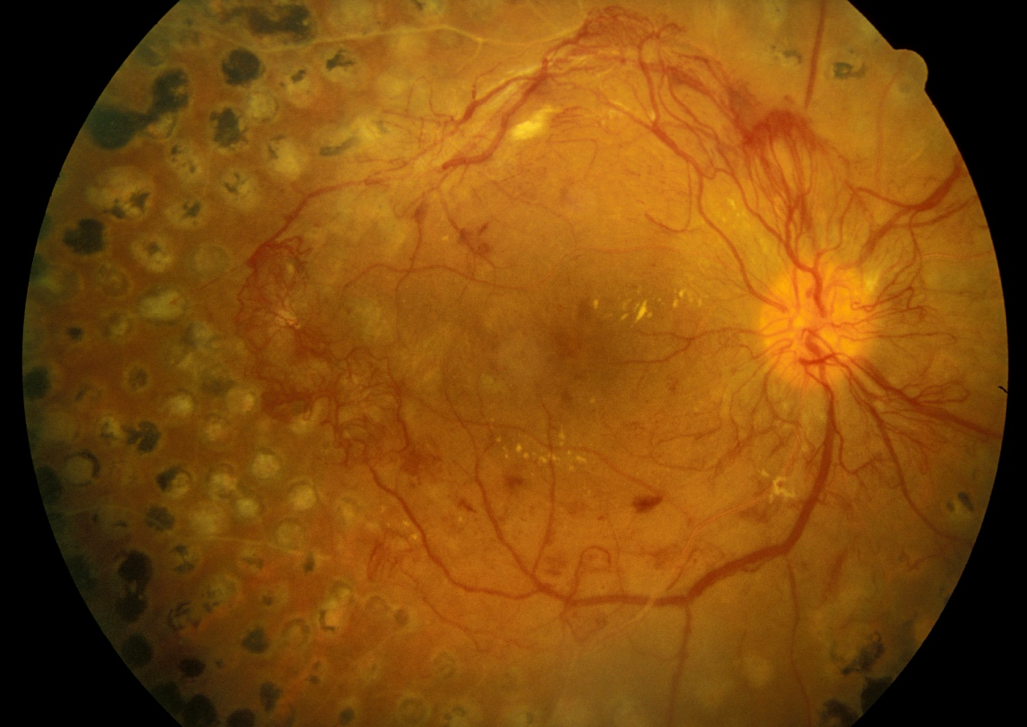
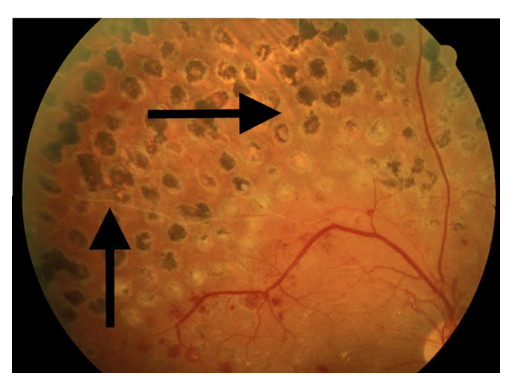
- Somasundaram NP, Ranathunga I, Ratnasamy V, Wijewickrama PSA, Dissanayake HA, Yogendranathan N, et al. The Impact of SARS-Cov-2 Virus Infection on the Endocrine System. Journal of the Endocrine Society. 2020;4(8).
- Pekic S, Miljic D, Popovic V. Infections of the Hypothalamic-Pituitary Region.Endotext [Internet]: MDText. com, Inc.; 2018.
- Aita K, Shiga J. Herpes simplex virus types 1 and 2 infect the mouse pituitary gland and induce apoptotic cell death. Archives of virology. 2004;149(12):2443-51.
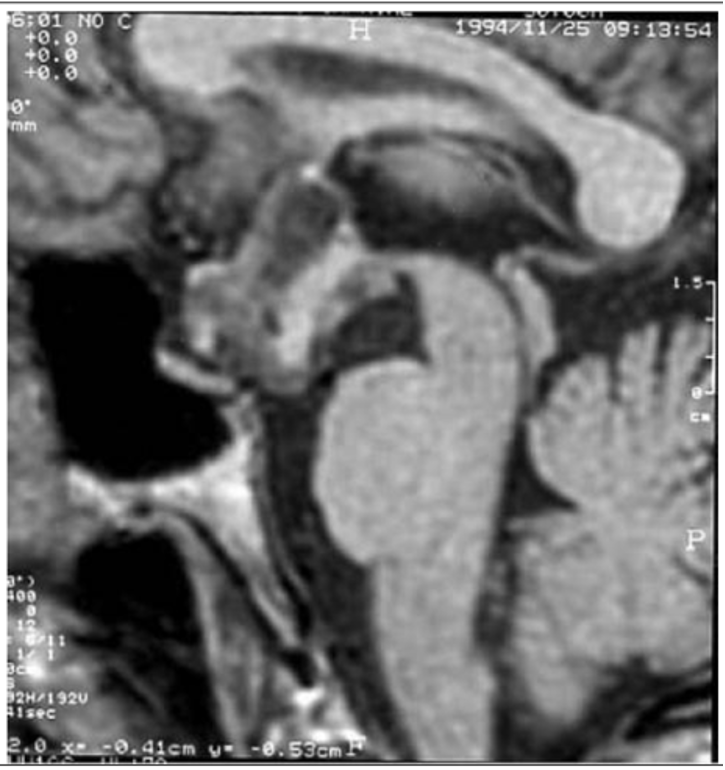
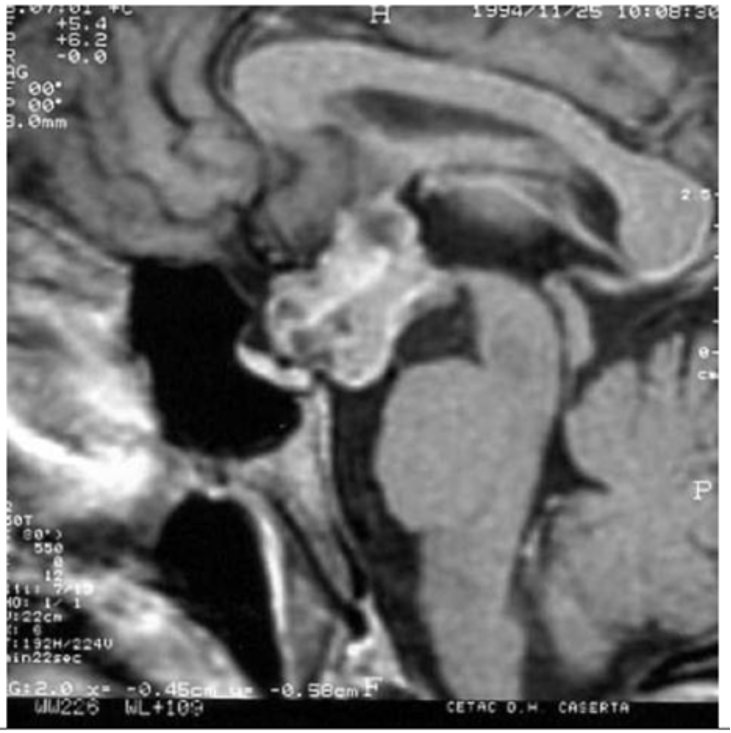
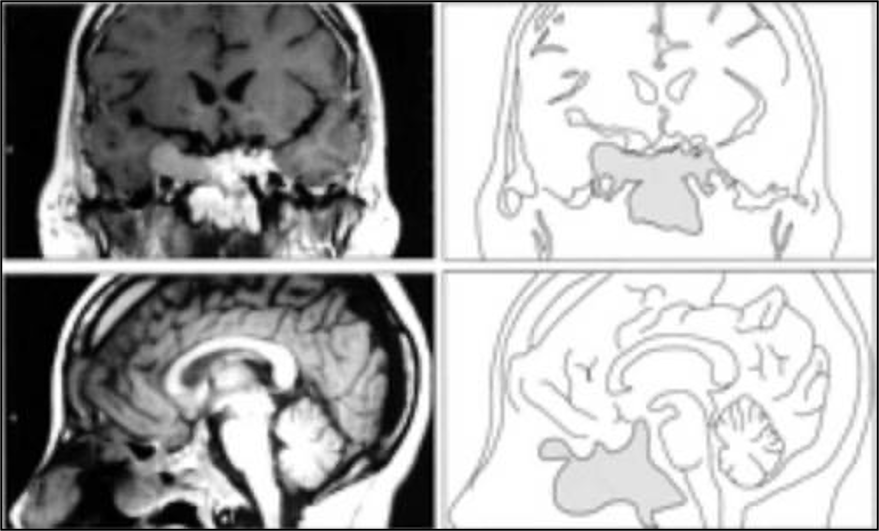
- Hautala T, Sironen T, Vapalahti O, Pääkkö E, Särkioja T, Salmela PI, et al. Hypophyseal hemorrhage and panhypopituitarism during Puumala Virus Infection: Magnetic Resonance Imaging and detection of viral antigen in the hypophysis. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2002;35(1):96-101.
- Sellmeyer DE, Grunfeld C. Endocrine and metabolic disturbances in human immunodeficiency virus infection and the acquired immune deficiency syndrome. Endocrine reviews. 1996;17(5):518-32.
- Yousuf T L, L. Bacal, A. Krikorian, A. A novel association between pituitary tumor apoplexy and influenza A. AACE Clinical Case Rep 2016. 2016;2:e143-e5.
- Suh DC, Park JS, Park SK, Lee HK, Chang KH. Pituitary hemorrhage as a complication of hantaviral disease. AJNR American journal of neuroradiology. 1995;16(1):175-8; discussion 9-80.
- Figurnov VA, Marunich NA, Gavrilov AV, Figurnova EV. [Late consequences of hemorrhagic fever with renal syndrome in a woman who had it during pregnancy]. Klinicheskaia meditsina. 2007;85(5):71-2.
- Raj H, Kamalanathan S, Sahoo JP, Kadhiravan T. Varicella causing remission of Cushing's disease. BMJ case reports. 2018;2018:bcr2018225867.
- Pal R. COVID-19, hypothalamo-pituitary-adrenal axis and clinical implications. Endocrine. 2020;68(2):251-2.
- Yoon JW, Choi DS, Liang HC, Baek HS, Ko IY, Jun HS, et al. Induction of an organ-specific autoimmune disease, lymphocytic hypophysitis, in hamsters by recombinant rubella virus glycoprotein and prevention of disease by neonatal thymectomy. J Virol. 1992;66(2):1210-4.
- Hägg E, Aström L, Steen L. Persistent hypothalamic-pituitary insufficiency following acute meningoencephalitis. A report of two cases. Acta medica Scandinavica. 1978;203(3):231-5.
- Kupari M, Pelkonen R, Valtonen V. Post-encephalitic hypothalamic-pituitary insufficiency. Acta endocrinologica. 1980;94(4):433-8.
- Lim AKH, Paramaswaran S, Jellie LJ, Junckerstorff RK. A Cross-Sectional Study of Hyponatremia Associated with Acute Central Nervous System Infections. Journal of Clinical Medicine. 2019;8(11):1801.
- Grajecki DS, Haas CS, Hubold C, Lehnert H. Sucessfull treatment of EBV-associated SIADH with V2 receptor antagonist. Exp Clin Endocrinol Diabetes. 2013;121(03):P68.
- Kuriki A, Ishihara K, Satoh H, Sugie M, Kato H, Kawamura M. [Syndrome of inappropriate secretion of anti-diuretic hormone associated with limbic encephalitis due to herpes simplex virus infection--a case report]. Rinsho shinkeigaku = Clinical neurology. 2008;48(3):184-90.
- Ahn HJ, Chung JH, Kim DM, Yoon NR, Kim CM. Hemorrhagic fever with renal syndrome accompanied by panhypopituitarism and central diabetes insipidus: a case report. Journal of neurovirology. 2018;24(3):382-7.
- Moses AM, Thomas DG, Canfield MC, Collins GH. Central Diabetes Insipidus due to Cytomegalovirus Infection of the Hypothalamus in a Patient with Acquired Immunodeficiency Syndrome: A Clinical, Pathological, and Immunohistochemical Case Study. The Journal of Clinical Endocrinology & Metabolism. 2003;88(1):51-4.
- Lee YJ, Yang D, Shyur SD, Chiu NC. Neurogenic diabetes insipidus in a child with fatal Coxsackie virus B1 encephalitis. Journal of pediatric endocrinology & metabolism : JPEM. 1995;8(4):301-4.
- Madhoun ZT, DuBois DB, Rosenthal J, Findlay JC, Aron DC. Central diabetes insipidus: a complication of herpes simplex type 2 encephalitis in a patient with AIDS. Am J Med. 1991;90(5):658-9.
- Borba VV, Zandman-Goddard G, Shoenfeld Y. Prolactin and Autoimmunity. Frontiers in Immunology. 2018;9(73).
- Parra A, Ramírez-Peredo J, Larrea F, Cabrera V, Coutiño B, Torres I, et al. Decreased dopaminergic tone and increased basal bioactive prolactin in men with human immunodeficiency virus infection. Clin Endocrinol (Oxf). 2001;54(6):731-8.
- Ochoa-Amaya JE, Malucelli BE, Cruz-Casallas PE, Nasello AG, Felicio LF, Carvalho-Freitas MI. Acute and chronic stress and the inflammatory response in hyperprolactinemic rats. Neuroimmunomodulation. 2010;17(6):386-95.
- McGill F, Griffiths MJ, Bonnett LJ, Geretti AM, Michael BD, Beeching NJ, et al. Incidence, aetiology, and sequelae of viral meningitis in UK adults: a multicentre prospective observational cohort study. The Lancet Infectious Diseases. 2018;18(9):992-1003.
- Czupryna P, Moniuszko A, Garkowski A, Pancewicz S, Zajkowska J. Comparison of hyponatremia and SIADH frequency in patients with tick borne encephalitis and meningitis of other origin. Scandinavian Journal of Clinical and Laboratory Investigation. 2016;76(2):159-64.
- Swart RM, Hoorn EJ, Betjes MG, Zietse R. Hyponatremia and Inflammation: The Emerging Role of Interleukin-6 in Osmoregulation. Nephron Physiology. 2011;118(2):p45-p51.
- Scheinpflug K, Schalk E, Reschke K, Franke A, Mohren M. Diabetes insipidus due to herpes encephalitis in a patient with diffuse large cell lymphoma. A case report. Exp Clin Endocrinol Diabetes. 2006;114(1):31-4.
- Torres AM, Kazee AM, Simon H, Knudson PE, Weinstock RS. Central diabetes insipidus due to herpes simplex in a patient immunosuppressed by Cushing's syndrome. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2000;6(1):26-8.
- Zou R, Wu C, Zhang S, Wang G, Zhang Q, Yu B, et al. Euthyroid Sick Syndrome in Patients With COVID-19. Frontiers in Endocrinology. 2020;11(798).
- Jonklaas J, Bianco AC, Bauer AJ, Burman KD, Cappola AR, Celi FS, et al. Guidelines for the treatment of hypothyroidism: prepared by the american thyroid association task force on thyroid hormone replacement. Thyroid. 2014;24(12):1670-751.
- Kojima M, Nakamura S, Oyama T, Sugihara S, Sakata N, Masawa N. Cellular composition of subacute thyroiditis. an immunohistochemical study of six cases. Pathology, research and practice. 2002;198(12):833-7.
- Eylan E, Zmucky R, Sheba C. Mumps virus and subacute thyroiditis; evidence of a causal association. Lancet (London, England). 1957;272(6978):1062-3.
- VOLPÉ R, ROW VV, EZRIN C. Circulating Viral and Thyroid Antibodies in Subacute Thyroiditis1. The Journal of Clinical Endocrinology & Metabolism. 1967;27(9):1275-84.
- Volpé R. The management of subacute (DeQuervain's) thyroiditis. Thyroid. 1993;3(3):253-5.
- Desailloud R, Hober D. Viruses and thyroiditis: an update. Virol J. 2009;6:5-.
- Dumoulin F, Liese H. Acute hepatitis E virus infection and autoimmune thyroiditis: yet another trigger? BMJ case reports. 2012;2012.
- Mori K, Yoshida K. Viral infection in induction of Hashimoto's thyroiditis: A key player or just a bystander? Current opinion in endocrinology, diabetes, and obesity. 2010;17:418-24.
- Davies TF. Infection and Autoimmune Thyroid Disease. The Journal of Clinical Endocrinology & Metabolism. 2008;93(3):674-6.
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annual review of immunology. 2003;21:335-76.
- Burch HB, Nagy EV, Lukes YG, Cai WY, Wartofsky L, Burman KD. Nucleotide and amino acid homology between the human thyrotropin receptor and the HIV-1 Nef protein: identification and functional analysis. Biochemical and biophysical research communications. 1991;181(1):498-505.
- Akahori H, Takeshita Y, Saito R, Kaneko S, Takamura T. Graves' Disease Associated with Infectious Mononucleosis due to Primary Epstein-Barr Virus Infection: Report of 3 Cases. Internal medicine (Tokyo, Japan). 2010;49:2599-603.
- Wang W, Ye Y, Yao H, Sun L, Wang A, Wang Z. Evaluation and observation of serum thyroid hormone and parathyroid hormone in patients with severe acute respiratory syndrome. J Chin Antituberculous Assoc. 2003;25:232-4.
- Ciampolillo A, Marini V, Mirakian R, Buscema M, Schulz T, Pujol-Borrell R, et al. Retrovirus-like sequences in Graves' disease: implications for human autoimmunity. Lancet (London, England). 1989;1(8647):1096-100.
- Mulcahy HEM, F.E. Freaney, R. Hegarty J.E. Parathyroid crisis and acute viral hepatitis B infection. Postgrad Med J. 1993;69:408-10.
- Hellman P, Albert J, Gidlund M, Klareskog L, Rastad J, Akerström G, et al. Impaired parathyroid hormone release in human immunodeficiency virus infection. AIDS research and human retroviruses. 1994;10(4):391-4.
- Elkattawy SA, R. Ayad, S. A Novel Case of Hypoparathyroidism Secondary to SARS-CoV-2 Infection. Cureus 2020;12(8):e10097.
- Sandhu S, Desai A, Batra M, Girdhar R, Chatterjee K, Kemp EH, et al. Severe Symptomatic Hypocalcemia from HIV Related Hypoparathyroidism. Case Reports in Endocrinology. 2018;2018:8270936.
- Singh A, Dnyanesh D. The prevalence of hypocalcemia in children with dengue infection: A 1-year cross-sectional study. Indian Journal of Health Sciences and Biomedical Research. 2019;12:166.
- Sobel R, Einhorn S, Maislos M, Shainkin-Kestenbaum R, Shany S, Horowitz J. Transitory Hypocalcemia Complicating Measles. Archives of Internal Medicine. 1985;145(11):2043-4.
- Jia-Kui S, Wen-Hao Z, Lei Z, Ying L, Jing-Jing L, Xiao-Hua K, et al. Research Square. 2021.
- Liu J, Han P, Wu J, Gong J, Tian D. Prevalence and predictive value of hypocalcemia in severe COVID-19 patients. Journal of Infection and Public Health. 2020;13(9):1224-8.
- Antonelli A, Ferrari SM, Di Domenicantonio A, Ferrannini E, Fallahi P. Chapter 53 - Viral Infections and Type 1 Diabetes. In: Shoenfeld Y, Agmon-Levin N, Rose NR, editors. Infection and Autoimmunity (Second Edition). Amsterdam: Academic Press; 2015. p. 877-89.
- Masini M, Campani D, Boggi U, Menicagli M, Funel N, Pollera M, et al. Hepatitis C Virus Infection and Human Pancreatic β-Cell Dysfunction. Diabetes Care. 2005;28(4):940-1.
- Capua I, Mercalli A, Pizzuto MS, Romero-Tejeda A, Kasloff S, De Battisti C, et al. Influenza A Viruses Grow in Human Pancreatic Cells and Cause Pancreatitis and Diabetes in an Animal Model. Journal of Virology. 2013;87(1):597-610.
- Sarmiento L, Frisk G, Anagandula M, Hodik M, Barchetta I, Netanyah E, et al. Echovirus 6 Infects Human Exocrine and Endocrine Pancreatic Cells and Induces Pro-Inflammatory Innate Immune Response. Viruses. 2017;9(2):25.
- Ginsberg-Fellner F, Witt ME, Fedun B, Taub F, Dobersen MJ, McEvoy RC, et al. Diabetes mellitus and autoimmunity in patients with the congenital rubella syndrome. Reviews of infectious diseases. 1985;7 Suppl 1:S170-6.
- Rawla P, Bandaru SS, Vellipuram AR. Review of Infectious Etiology of Acute Pancreatitis. Gastroenterology Res. 2017;10(3):153-8.
- Zhi M, Zhu X, Lugea A, Waldron RT, Pandol SJ, Li L. Incidence of New Onset Diabetes Mellitus Secondary to Acute Pancreatitis: A Systematic Review and Meta-Analysis. Front Physiol. 2019;10:637-.
- Boettler T, von Herrath M. Protection against or triggering of Type 1 diabetes? Different roles for viral infections. Expert Rev Clin Immunol. 2011;7(1):45-53.
- Christen U, von Herrath MG. Do viral infections protect from or enhance type 1 diabetes and how can we tell the difference? Cellular & Molecular Immunology. 2011;8(3):193-8.
- Zhao G, Vatanen T, Droit L, Park A, Kostic AD, Poon TW, et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proceedings of the National Academy of Sciences. 2017;114(30):E6166-E75.
- Pusch E, Renz H, Skevaki C. Respiratory virus-induced heterologous immunity. Allergo Journal. 2018;27(3):28-45.
- Paolo WF, Nosanchuk JD. Adrenal infections. International Journal of Infectious Diseases. 2006;10(5):343-53.
- Potratz D, Brake B, Dienes H, Schulz TF, Hosp M, Dierich M, et al. Herpes simplex virus type 1 and 2 in the adrenal glands: replication and histopathology. Archives of virology. 1986;90(3-4):207-22.
- Ding Y, He L, Zhang Q, Huang Z, Che X, Hou J, et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS‐CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland. 2004;203(2):622-30.
- Wheatland R. Molecular mimicry of ACTH in SARS–implications for corticosteroid treatment and prophylaxis. Medical hypotheses. 2004;63(5):855-62.
- Lee C-W, Suarez DL, Tumpey TM, Sung H-W, Kwon Y-K, Lee Y-J, et al. Characterization of highly pathogenic H5N1 avian influenza A viruses isolated from South Korea. Journal of virology. 2005;79(6):3692-702.
- Mahanty S, Bray M. Pathogenesis of filoviral haemorrhagic fevers. The Lancet infectious diseases. 2004;4(8):487-98.
- Ventura KC, Hawkins H, Smith MB, Walker DH. Fatal neonatal echovirus 6 infection: autopsy case report and review of the literature. Modern Pathology. 2001;14(2):85-90.
- Pinching AJ. Cytomegalovirus infection in the acquired immune deficiency syndrome. Journal of Antimicrobial Chemotherapy. 1989;23(suppl_E):31-6.
- Glasgow BJ, Steinsapir KD, Anders K, Layfield LJ. Adrenal pathology in the acquired immune deficiency syndrome. American Journal of Clinical Pathology. 1985;84(5):594-7.
- Hofbauer LC, Heufelder AE. Endocrine implications of human immunodeficiency virus infection. Medicine. 1996;75(5):262-78.
- Welch K, Finkbeiner W, Alpers CE, Blumenfeld W, Davis RL, Smuckler EA, et al. Autopsy findings in the acquired immune deficiency syndrome. Jama. 1984;252(9):1152-9.
- Salim YS, Faber V, Wiik A, Andersen PL, HØIer‐Madsen M, Mouritsen S. Anti‐corticosteroid antibodies in AIDS patients. Apmis. 1988;96(7‐12):889-94.
- Eledrisi MS, Verghese AC. Adrenal insufficiency in HIV infection: a review and recommendations. The American journal of the medical sciences. 2001;321(2):137-44.
- Marik PE, Kiminyo K, Zaloga GP. Adrenal insufficiency in critically ill patients with human immunodeficiency virus. Critical care medicine. 2002;30(6):1267-73.
- Rodrigues D, Reis M, Teixeira V, Silva-Vergara M, Correia Filho D, Adad S, et al. Pathologic findings in the adrenal glands of autopsied patients with acquired immunodeficiency syndrome. Pathology-Research and Practice. 2002;198(1):25-30.
- Ohsawa M, Tomita Y, Hashimoto M, Yasunaga Y, Kanno H, Aozasa K. Malignant lymphoma of the adrenal gland: its possible correlation with the Epstein-Barr virus. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 1996;9(5):534.
- Masarani M, Wazait H, Dinneen M. Mumps orchitis. J R Soc Med. 2006;99(11):573-5.
- Barták V. Sperm count, morphology and motility after unilateral mumps orchitis. Journal of reproduction and fertility. 1973;32(3):491-4.
- Wu H, Shi L, Wang Q, Cheng L, Zhao X, Chen Q, et al. Mumps virus-induced innate immune responses in mouse Sertoli and Leydig cells. Scientific Reports. 2016;6(1):19507.
- Casella R, Leibundgut B, Lehmann K, Gasser TC. Mumps orchitis: report of a mini-epidemic. The Journal of urology. 1997;158(6):2158-61.
- Adamopoulos DA, Lawrence DM, Vassilopoulos P, Contoyiannis PA, Swyer GI. Pituitary-testicular interrelationships in mumps orchitis and other viral infections. Br Med J. 1978;1(6121):1177-80.
- Craighead JE. CHAPTER 1 - Enteroviruses. In: Craighead JE, editor. Pathology and Pathogenesis of Human Viral Disease. San Diego: Academic Press; 2000. p. 1-28.
- Matusali G, Houzet L, Satie A-P, Mahé D, Aubry F, Couderc T, et al. Zika virus infects human testicular tissue and germ cells. The Journal of Clinical Investigation. 2018;128(10):4697-710.
- Huang JM, Huang TH, Qiu HY, Fang XW, Zhuang TG, Qiu JW. Studies on the integration of hepatitis B virus DNA sequence in human sperm chromosomes. Asian journal of andrology. 2002;4(3):209-12.
- Joguet G MJM, Matusali G, Hamdi S, Walschaerts M, Pavili L, Guyomard S, Prisant N, Lamarre P, Dejucq-Rainsford N, Pasquier C, Bujan L. Effect of acute Zika virus infection on sperm and virus clearance in body fluids: a prospective observational study. The Lancet Infectious Diseases. 2017;17 (11):1200-8.
- Feldmann H. Virus in Semen and the Risk of Sexual Transmission. New England Journal of Medicine. 2018;378(15):1440-1.
- Li D, Jin M, Bao P, Zhao W, Zhang S. Clinical Characteristics and Results of Semen Tests Among Men With Coronavirus Disease 2019. JAMA Network Open. 2020;3(5):e208292-e.
- Le Tortorec A, Satie A-P, Denis H, Rioux-Leclercq N, Havard L, Ruffault A, et al. Human prostate supports more efficient replication of HIV-1 R5 than X4 strains ex vivo. Retrovirology. 2008;5(1):119.
- Willey S, Roulet V, Reeves JD, Kergadallan M-L, Thomas E, McKnight Á, et al. Human Leydig cells are productively infected by some HIV-2 and SIV strains but not by HIV-1. Aids. 2003;17(2):183-8.
- Yuan J. Genitourinary Presentation of Tuberculosis. Reviews in urology. 2015;17(2):102-5.
- Garolla A, Vitagliano A, Muscianisi F, Valente U, Ghezzi M, Andrisani A, et al. Role of Viral Infections in Testicular Cancer Etiology: Evidence From a Systematic Review and Meta-Analysis. Frontiers in endocrinology. 2019;10:355-.
- Naz RK, Ellaurie M, Phillips TM, Hall J. Antisperm antibodies in human immunodeficiency virus infection: effects on fertilization and embryonic development. Biology of reproduction. 1990;42(5-6):859-68.
- Morrison JC, Givens JR, Wiser WL, Fish SA. Mumps oophoritis: a cause of premature menopause. Fertility and sterility. 1975;26(7):655-9.
- Morelli F, Souza RP, Cruz TEd, Damke GMZF, Damke E, Suehiro TT, et al. Zika virus infection in the genital tract of non-pregnant females: a systematic review. Revista do Instituto de Medicina Tropical de São Paulo. 2020;62.
- Manfredi R, Alampi G, Talò S, Calza L, Tadolini M, Martinelli GN, et al. Silent oophoritis due to cytomegalovirus in a patient with advanced HIV disease. International journal of STD & AIDS. 2000;11(6):410-2.
- Ingerslev K, Hogdall E, Schnack TH, Skovrider-Ruminski W, Hogdall C, Blaakaer J. The potential role of infectious agents and pelvic inflammatory disease in ovarian carcinogenesis. Infectious Agents and Cancer. 2017;12(1):25.
- Altindis E, Cai W, Sakaguchi M, Zhang F, GuoXiao W, Liu F, et al. Viral insulin-like peptides activate human insulin and IGF-1 receptor signaling: A paradigm shift for host–microbe interactions. Proceedings of the National Academy of Sciences. 2018;115(10):2461-6.
- Irwin DM. Viral Insulin/IGF-1–Like Peptides: Novel Regulators of Physiology and Pathophysiology? Endocrinology. 2018;159(11):3659-60.
- Huang Q, Kahn CR, Altindis E. Viral Hormones: Expanding Dimensions in Endocrinology. Endocrinology. 2019;160(9):2165-79.
- Watanabe T, Yamaguchi K, Takatsuki K, Osame M, Yoshida M. Constitutive expression of parathyroid hormone-related protein gene in human T cell leukemia virus type 1 (HTLV-1) carriers and adult T cell leukemia patients that can be trans-activated by HTLV-1 tax gene. The Journal of experimental medicine. 1990;172(3):759-65.
- Kino T, De Martino MU, Charmandari E, Mirani M, Chrousos GP. Tissue glucocorticoid resistance/hypersensitivity syndromes. The Journal of steroid biochemistry and molecular biology. 2003;85(2-5):457-67.
- Roodman GD, Windle JJ. Paget disease of bone. The Journal of clinical investigation. 2005;115(2):200-8.
- Glantschnig C, Koenen M, Lozano MG, Karbiener M, Cummins C, Blüher M, et al. A miR-29a-driven negative feedback loop regulates the glucocorticoid receptor in health and disease. Diabetologie und Stoffwechsel. 2018;13(03):LB FV 7.
- Chen N, Baudino T, MacDonald PN, Green M, Kelley WL, Burnett JW, et al. Selective inhibition of nuclear steroid receptor function by a protein from a human tumorigenic poxvirus. Virology. 2000;274(1):17-25.
- Brockmann D, Esche H. The multifunctional role of E1A in the transcriptional regulation of CREB/CBP-dependent target genes. Current topics in microbiology and immunology. 2003;272:97-129.
- Puig-Domingo M, Marazuela M, Giustina A. COVID-19 and endocrine diseases. A statement from the European Society of Endocrinology. Endocrine. 2020;68(1):2-5.
- Bergthorsdottir R, Leonsson-Zachrisson M, Odén A, Johannsson G. Premature mortality in patients with Addison’s disease: a population-based study. The Journal of Clinical Endocrinology & Metabolism. 2006;91(12):4849-53.
- Quinkler M, Ekman B, Zhang P, Isidori AM, Murray RD, Investigators EA. Mortality data from the European Adrenal Insufficiency Registry—Patient characterization and associations. Clinical endocrinology. 2018;89(1):30-5.
- Tresoldi AS, Sumilo D, Perrins M, Toulis KA, Prete A, Reddy N, et al. Increased infection risk in Addison’s disease and congenital adrenal hyperplasia: a primary care database cohort study. The Journal of Clinical Endocrinology & Metabolism. 2020;105(2):418-29.
- Bancos I, Hazeldine J, Chortis V, Hampson P, Taylor AE, Lord JM, et al. Primary adrenal insufficiency is associated with impaired natural killer cell function: a potential link to increased mortality. European journal of endocrinology. 2017;176(4):471.
- Isidori AM, Venneri MA, Graziadio C, Simeoli C, Fiore D, Hasenmajer V, et al. Effect of once-daily, modified-release hydrocortisone versus standard glucocorticoid therapy on metabolism and innate immunity in patients with adrenal insufficiency (DREAM): a single-blind, randomised controlled trial. The lancet Diabetes & endocrinology. 2018;6(3):173-85.
- Isidori AM, Arnaldi G, Boscaro M, Falorni A, Giordano C, Giordano R, et al. COVID-19 infection and glucocorticoids: update from the Italian Society of Endocrinology Expert Opinion on steroid replacement in adrenal insufficiency. Journal of endocrinological investigation. 2020;43(8):1141-7.
- Ives AM, Bertke AS. Stress Hormones Epinephrine and Corticosterone Selectively Modulate Herpes Simplex Virus 1 (HSV-1) and HSV-2 Productive Infections in Adult Sympathetic, but Not Sensory, Neurons. Journal of Virology. 2017;91(13):e00582-17.
- Noisakran S, Halford WP, Veress L, Carr DJJ. Role of the Hypothalamic Pituitary Adrenal Axis and IL-6 in Stress-Induced Reactivation of Latent Herpes Simplex Virus Type 1. The Journal of Immunology. 1998;160(11):5441-7.
- Uchakin PN, Parish DC, Dane FC, Uchakina ON, Scheetz AP, Agarwal NK, et al. Fatigue in Medical Residents Leads to Reactivation of Herpes Virus Latency. Interdisciplinary Perspectives on Infectious Diseases. 2011;2011:571340.
- Englund J, Feuchtinger T, Ljungman P. Viral infections in immunocompromised patients. Biol Blood Marrow Transplant. 2011;17(1 Suppl):S2-S5.
- Pivonello R, Ferrigno R, Isidori AM, Biller BMK, Grossman AB, Colao A. COVID-19 and Cushing's syndrome: recommendations for a special population with endogenous glucocorticoid excess. Lancet Diabetes Endocrinol. 2020;8(8):654-6.
- Pivonello R, Isidori AM, De Martino MC, Newell-Price J, Biller BM, Colao A. Complications of Cushing's syndrome: state of the art. The Lancet Diabetes & Endocrinology. 2016;4(7):611-29.
- Berczi I. Pituitary hormones and immune function. Acta Paediatrica. 1997;86(S423):70-5.
- Postel-Vinay M-C, de Mello Coelho V, Gagnerault M-C, Dardenne M. Growth hormone stimulates the proliferation of activated mouse T lymphocytes. Endocrinology. 1997;138(5):1816-20.
- Yu-Lee L-Y. Molecular actions of prolactin in the immune system. Proceedings of the Society for Experimental Biology and Medicine. 1997;215(1):35-52.
- Fabris N, Mocchegiani E, Provinciali M. Pituitary-Thyroid Axis and Immune System: A Reciprocal Neuroendocrine-lmmune Interaction. Hormone Research in Paediatrics. 1995;43(1-3):29-38.
- Grossman CJ. Interactions between the gonadal steroids and the immune system. Science. 1985;227(4684):257-61.
- Sharma MD, Sagar B, Wang S, Clinton White A, Jr., Balasubramanyam A. High Frequency of Serious Infections in Patients with Panhypopituitarism: A Case-Control Study. Clinical Infectious Diseases. 2001;32(1):153-8.
- Bialasiewicz AA, Jüppner H, Diehl V, Hesch R-D. Binding of bovine parathyroid hormone to surface receptors of cultured B-lymphocytes. Biochimica et Biophysica Acta (BBA)-General Subjects. 1979;584(3):467-78.
- Shurtz-Swirski R, Shkolnik T, Shasha SM. Parathyroid hormone and the cellular immune system. Nephron. 1995;70(1):21-4.
- El Kelany MA, Maklad SS, El Fouhil DF, El Etreby EA, Saleh LH, El Sheikh NA. Immunomodulatory effects of secondary hyperparathyroidism on circulating CD4+ and CD8+ T-lymphocytes in chronic renal failure patients. The Egyptian journal of immunology. 2009;16(2):71-82.
- Klein SL. Sex influences immune responses to viruses, and efficacy of prophylaxis and treatments for viral diseases. Bioessays. 2012;34(12):1050-9.
- Klein SL, Jedlicka A, Pekosz A. The Xs and Y of immune responses to viral vaccines. The Lancet infectious diseases. 2010;10(5):338-49.
- Hewagama A, Patel D, Yarlagadda S, Strickland FM, Richardson BC. Stronger inflammatory/cytotoxic T-cell response in women identified by microarray analysis. Genes & Immunity. 2009;10(5):509-16.
- Kovats S, Carreras E, Agrawal H, Klein S, Roberts C. Sex hormones and immunity to infection. 2010.
- Tuku B, Stanelle-Bertram S, Sellau J, Beck S, Bai T, Kouassi NM, et al. Testosterone Protects Against Severe Influenza by Reducing the Pro-Inflammatory Cytokine Response in the Murine Lung. Frontiers in Immunology. 2020;11(697).
- Steeg LGv, Vermillion MS, Hall OJ, Alam O, McFarland R, Chen H, et al. Age and testosterone mediate influenza pathogenesis in male mice. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2016;311(6):L1234-L44.
- vom Steeg LG, Dhakal S, Woldetsadik YA, Park H-S, Mulka KR, Reilly EC, et al. Androgen receptor signaling in the lungs mitigates inflammation and improves the outcome of influenza in mice. PLOS Pathogens. 2020;16(7):e1008506.
- Robinson DP, Hall OJ, Nilles TL, Bream JH, Klein SL. 17β-estradiol protects females against influenza by recruiting neutrophils and increasing virus-specific CD8 T cell responses in the lungs. Journal of virology. 2014;88(9):4711-20.
- Straub RH. The complex role of estrogens in inflammation. Endocrine reviews. 2007;28(5):521-74.
- Costeira R, Lee KA, Murray B, Christiansen C, Castillo-Fernandez J, Ni Lochlainn M, et al. Estrogen and COVID-19 symptoms: associations in women from the COVID Symptom Study. medRxiv. 2020:2020.07.30.20164921.
- Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus (DM). FEMS immunology and medical microbiology. 1999;26(3-4):259-65.
- Muller LM, Gorter KJ, Hak E, Goudzwaard WL, Schellevis FG, Hoepelman AI, et al. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2005;41(3):281-8.
- Diepersloot R, Bouter KP, Beyer W, Hoekstra J, Masurel N. Humoral immune response and delayed type hypersensitivity to influenza vaccine in patients with diabetes mellitus. Diabetologia. 1987;30(6):397-401.
- Singh AK, Gupta R, Ghosh A, Misra A. Diabetes in COVID-19: Prevalence, pathophysiology, prognosis and practical considerations. Diabetes & Metabolic Syndrome: Clinical Research & Reviews. 2020.
- Tong Q, Xiang W, Ye H, Zhang H, Chen Y, Chen W, et al. T3 level may be a helpful marker to predict disease prognosis of acute central nervous system viral infections. The International journal of neuroscience. 2019;129(2):139-45.
- Constantine G, Rajapakse S, Ranasinghe P, Parththipan B, Wijewickrama A, Jayawardana P. Hypocalcemia is associated with disease severity in patients with dengue. Journal of infection in developing countries. 2014;8:1205-9.
- Manjunath VG, Balla S, Kumar J. Serum ionic calcium levels and hypocalcemia in dengue fever in children and its correlation with its severity: case control study. 2019. 2019;6(3):5.
- Jin Y, Yang H, Ji W, Wu W, Chen S, Zhang W, et al. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses. 2020;12(4):372.
- Lazartigues E, Qadir MMF, Mauvais-Jarvis F. Endocrine Significance of SARS-CoV-2’s Reliance on ACE2. Endocrinology. 2020;161(9).
- Zinserling VA, Semenova NY, Markov AG, Rybalchenko OV, Wang J, Rodionov RN, et al. Inflammatory Cell Infiltration of Adrenals in COVID-19. Horm Metab Res. 2020;52(09):639-41.
- Pascual-Goñi E, Fortea J, Martínez-Domeño A, Rabella N, Tecame M, Gómez-Oliva C, et al. COVID-19-associated ophthalmoparesis and hypothalamic involvement. Neurology - Neuroimmunology Neuroinflammation. 2020;7(5):e823.
- Nampoothiri S, Sauve F, Ternier G, Fernandois D, Coelho C, Imbernon M, et al. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis. bioRxiv. 2020:2020.06.08.139329.
- Chen M, Zhou W, Xu W. Thyroid function analysis in 50 patients with COVID-19: a retrospective study. Thyroid. 2020.
- Rajevac H, Bachan M, Khan Z. DIABETES INSIPIDUS AS A SYMPTOM OF COVID-19 INFECTION: CASE REPORT. Chest. 2020;158(4):A2576-A.
- Yousaf Z, Al-Shokri SD, Al-Soub H, Mohamed MFH. COVID-19-associated SIADH: a clue in the times of pandemic! Am J Physiol Endocrinol Metab. 2020;318(6):E882-E5.
- Uddin Chowdhury MR, Akter KS, Moula MM, Kabir MA, Bhuiyan SI, Das BC. COVID-19 presented with syndrome of inappropriate ADH secretion(SIADH): A case report from Bangladesh. Respiratory Medicine Case Reports. 2020;31:101290.
- Ravioli S, Niebuhr N, Ruchti C, Pluess E, Stoeckli T, Lindner G. The syndrome of inappropriate antidiuresis in COVID-19 pneumonia: report of two cases. Clinical Kidney Journal. 2020.
- Brancatella A, Ricci D, Cappellani D, Viola N, Sgrò D, Santini F, et al. Is Subacute Thyroiditis an Underestimated Manifestation of SARS-CoV-2 Infection? Insights From a Case Series. The Journal of Clinical Endocrinology & Metabolism. 2020;105(10):e3742-e6.
- Brancatella A, Ricci D, Viola N, Sgrò D, Santini F, Latrofa F. Subacute thyroiditis after SARS-CoV-2 infection. The Journal of Clinical Endocrinology & Metabolism. 2020;105(7):dgaa276.
- Mateu-Salat M, Urgell E, Chico A. SARS-COV-2 as a trigger for autoimmune disease: report of two cases of Graves’ disease after COVID-19. Journal of endocrinological investigation. 2020;43(10):1527-8.
- Muller I, Cannavaro D, Dazzi D, Covelli D, Mantovani G, Muscatello A, et al. SARS-CoV-2-related atypical thyroiditis. The Lancet Diabetes & Endocrinology. 2020;8(9):739-41.
- Ceriello A, De Nigris V, Prattichizzo F. Why is hyperglycaemia worsening COVID‐19 and its prognosis? Diabetes, Obesity and Metabolism. 2020;22(10):1951-2.
- Yang J-K, Lin S-S, Ji X-J, Guo L-M. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta diabetologica. 2010;47(3):193-9.
- Szatmary P, Arora A, Raraty MGT, Dunne DFJ, Baron RD, Halloran CM. Emerging phenotype of severe acute respiratory syndrome-coronavirus 2–associated pancreatitis. Gastroenterology. 2020;159(4):1551-4.
- Reddy PK, Kuchay MS, Mehta Y, Mishra SK. Diabetic ketoacidosis precipitated by COVID-19: A report of two cases and review of literature. Diabetes Metab Syndr. 2020;14(5):1459-62.
- Iuga AC, Marboe CC, M. Yilmaz M, Lefkowitch JH, Gauran C, Lagana SM. Adrenal Vascular Changes in COVID-19 Autopsies. Archives of Pathology & Laboratory Medicine. 2020;144(10):1159-60.
- Frankel M, Feldman I, Levine M, Frank Y, Bogot NR, Benjaminov O, et al. Bilateral Adrenal Hemorrhage in Coronavirus Disease 2019 Patient: A Case Report. The Journal of Clinical Endocrinology & Metabolism. 2020;105(12):3745-9.
- Gagliardi L, Bertacca C, Centenari C, Merusi I, Parolo E, Ragazzo V, et al. Orchiepididymitis in a boy with COVID-19. The Pediatric infectious disease journal. 2020;39(8):e200-e2.
- Illiano E, Trama F, Costantini E. Could COVID‐19 have an impact on male fertility? Andrologia. 2020:e13654.
- Ma L, Xie W, Li D, Shi L, Mao Y, Xiong Y, et al. Effect of SARS-CoV-2 infection upon male gonadal function: A single center-based study. medRxiv. 2020:2020.03.21.20037267.
- Li H, Xiao X, Zhang J, Zafar MI, Wu C, Long Y, et al. Impaired spermatogenesis in COVID-19 patients. EClinicalMedicine. 2020;28.
- Serban AL, Ferrante E, Carosi G, Indirli R, Arosio M, Mantovani G. COVID-19 in Cushing disease: experience of a single tertiary centre in Lombardy. Journal of endocrinological investigation. 2020.
- Arlt W, Baldeweg SE, Pearce SH, Simpson HL. Clinical management guidance during the covid-19 pandemic adrenal insufficiency. European journal of endocrinology. 2020:1-21.