Incidence and Distribution
The annual incidence of thyroid cancer varies considerably in different registries, ranging from 1.2-2.6 per 100,000 individuals in men and from 2.0-3.8 per 100,000 in women (106,107). It is particularly elevated in Iceland and Hawaii, being nearly two times higher than in North European countries, Canada and the USA. In Hawaii, the incidence rate of thyroid cancer in each ethnic group is higher than that registered in their country of origin (108), and it is particularly common among Chinese males and Filipino females. Most of the differences are probably due to ethnic or environ¬mental factors (such as spontaneous background radiation) or dietary habits (109), but different standards of medical expertise and health care may also play a role in the efficiency of cancer detection. The American Cancer Society indicated incidence in the USA of nearly 10/100,000 population in 2003. The reported incidence has been increasing at more than 5%/yr for a decade.
In sharp contrast with these data concerning the incidence of clinical thyroid cancer, is the prevalence found in autopsy series or screening programs. Autopsy studies indicate a surprising frequency ranging from 0.01 to over 2.0% (110,111).A survey of consecutive autopsies at Grace-New Haven Hospital found 2.7% of thyroids to harbour unsuspected thyroid cancer (111). Another 2.7% had discrete benign adenomas, and nearly half showed nodularity. The high prevalence may be attributed to careful examination of the gland, but probably also reflects a highly selected group of older patients dying in a hospital. Up to 6% of thyroid glands in autopsied adults in the United States, and over 20% in Japan, also harbour microscopically detectable foci of thyroid carcinoma, which are believed to be of no biologic significance. Altogether autopsy studies suggest that thyroid cancer is in many instances not diagnosed dur¬ing life or is not the immediate cause of death. Both suggestions are in agreement with the rather leisurely growth of the majority of thyroid tumors, especially the frequent small papillary types.
The annual mortality from thyroid cancer in 2003 was 5 per million for men and 6 per million for women (112). The discrepancy between incidence and mortality reflects the good prognosis for most thyroid cancers. Recent statistics suggest about 6 deaths /million in the USA.
| Table 18-1. Neoplasms of the Thyroid(Adapted, and Revised, from WHO Classification) 12 |
A. Follicular
B. Papillary (probably malignant) C. Teratoma II. Malignant Tumors A. Differentiated
B. Medullary carcinoma C. Undifferentiated
D. Miscellaneous
|
Thyroid tumors are rare in children and increase in frequency in each decade. Carcinomas are two-three times as frequent in women as in men. In the past, it was generally believed that thyroid tumors were more frequent in areas of endemic goiter, and reports from Colombia and Austria support this association (113) (see Chapter 11). More recent studies suggest that in iodine deficient countries the number of nodules is increased and, as a consequence, also the number of thyroid cancers is increased (114). Surveys conducted in the United States found no relation between usual geographic residence and incidence of thyroid cancer.
ETIOLOGY
Most, if not all, thyroid adenomas are monoclonal, as, presumably, are most carcino¬mas (115). Colloid nodules may be either mono-or poly-clonal. Thus tumors represent the persistent growth of the progeny of one cell which has somehow escaped the mechanisms which maintain normal cell division at about once each 8.5 years (116).
The process of oncogenesis is conceived to be a series of events induced by genetic and environmental factors which alter growth control. At the phenomenologic level these factors may be considered as "initiators" and "promoters". Initiators include such agents as chemicals and irradiation which induce tumors, and promoters are agents such as phenobarbital, which in rats augments TSH secretion and radically increases tumor development. In man x-ray treatment is the sole known initiator, and other than elevated TSH, no promoters are known. Compounds such as phenobarbi¬tal, dilantin and PCBs, which are known thyroid tumor promoters in animals through liver microsomal hormone degrading enzyme induction leading to increased thyroid hormone metabolism, do not appear to have a detectable adverse effect in man in doses usually employed (117).
Oncogenes (Fig. 18-11)
We now begin to understand oncogenesis in more details. More than 30 "oncogenes" have been recognized in the human genome. The most likely genetic events in thyroid cancer are reported in Fig. 18-11. These genes, normally silent, can be¬come activated by chromosomal translocations, deletions, or mutations, and then can "transform" normal cells into a condition of uncontrolled growth. Most onco¬genes appear to be closely related to normal growth factors, genes that control cell division, or to hormone receptors. In general, these genes, when turned on, promote cell growth and cell division and depress differentiation. Typically activation of one such gene may not be enough to produce malignancy, but if accompanied by ex¬pression of another oncogene, or if gene mutation or reduplication occurs, the cell may progress toward a malignant potential. Information on expression of oncogenes in human thyroid tissue is rapidly accumulating. Expression of c-myc is stimulated in normal thyroid cells by TSH, and the proto-oncogene is expressed in adenomas and carcinomas. Activating mutations of h-ras at codons 12, 13, and 61, and over expression of h-ras, are found in adenomas and carcinomas, but h-ras mutations are also found in nodular goiter tissue (118), suggesting that h-ras mutations could be an early event in oncogenesis (119). Other studies, it should be noted, find ras mutations uncommon (120).
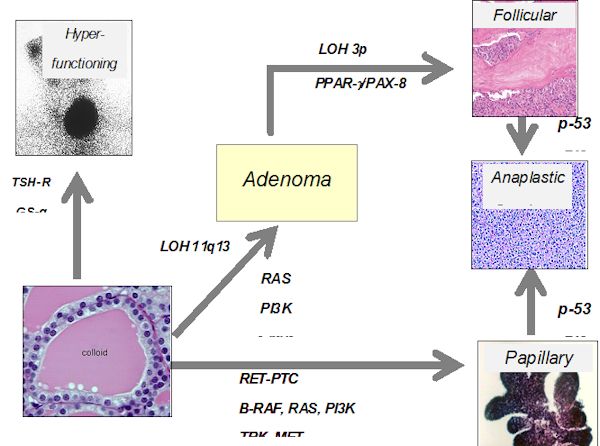
Figure 18-11. Possible role of oncogene activation, receptor or G-protein mutation, or tumor repressor gene alteration in the induction of thyroid carcinoma.
Santoro and co-workers (121) cloned an oncogene which is frequently and specifically expressed in papillary thyroid cancers. This oncogene is found on chromosome 10 and involves an intrachromosomal rearrangement of the tyrosine kinase domain of the ret oncogene so that it is attached to one of three different promoters, produc¬ing retPTC-1, retPTC-2, and retPTC-3. As a mean, one of these translocation products is found in about 20% of PTC, although in different series a large variation is observed (20-70%). This rearrangement leads to constitutive expression of the oncogene. It has been shown that intra-thyroidal expression of the ret/PTC1 oncogene can induce thyroid cancer (122). BRAF mutations, in the form of point mutations, are the most frequent alterations in papillary carcinoma, and undifferentiated cancers that have arisen from papillary tumors (123), approaching 40% of all PTCs (124).
Recently a mutational change has been associated with follicular cancers. In 5 of 8 follicular cancers, Kroll et al (125) found translocation of the DNA binding domain of PAX8 to domains A-F of the peroxisome proliferator-activater receptor (PPAR) gamma1 gene. The fusion oncogene is able to transform thyrocytes, so appears to be able to produce malignancies (126). Although initially thought to be exclusively present in follicular cancers, it is now known to be present in follicular adenomas as well (127).
Mutation or deletion of the p53 tumor suppressor gene is found in some differentiated thyroid cancers, and many undifferentiated cancers, suggesting that this genetic deletion may be one of the final steps leading to anaplastic cancer growth. A proliferation of studies in this field has provided many clues to thyroid tumorigenesis. Simian virus 40-like sequences are found in many thyroid cancers, as well as other cancers, and the Tag gene sequence found is known to be oncogenic in animal models (128). Mutated and non-functional thyroid hormone receptors are recognized in up to 90% of PTC by one author, suggesting a role in oncogenesis, but other workers find these mutations to be rare (129,130). The tumor suppressor gene TSG101 is over-expressed in most PTCs (131). Overexpression of many other genes -galectin-3, Thymosin beta-10,hTERT, CD97, CD26, VEGF-has been detected, but of course a question always is whether these changes represent the cause or the result of oncogenesis.
Mutations in the proteins involved in the normal TSH-receptor-G protein-adenyl¬cyclase-kinase signal transduction pathway also play a role in tumor formation. Activating TSH receptor mutations have been found by Parma and co-workers (132) to be the cause of most hyperfunctional nodules, and are now known to be common in "hot" nodules in patients with multi-nodular goiter.. These mutations involve the extracellular loops of the transmembrane domain and the transmembrane segments, and are proven to induce hyperfunction by transfection studies. However these mutations are not associated with cancer formation. Mutations of the stimulatory GTP binding protein subunit are also present in some patients with hyperfunctioning thyroid adenomas (133). TSH-R mutations are, however, unusual in thyroid cancer (134), (excepting hyperfunctional adenomas). TSH-R expression tends to be lost as cancers de-differentiate, and persistence of expression is associated with a better prognosis (135).
In addition to positive genetic factors, oncogenesis frequently involves loss of tumor suppressor genes. This has been proven in hereditary retinoblastoma. These genes are normally present on both sets (maternal and paternal) of chromosomes. In retinoblastoma the inherited lack of one suppressor (RB) gene does not cause disease, but if a genetic event (deletion, recombination, mutation, etc.) causes failure of expression of the second allele, cancer ensues. The presence of tumor-specific suppressor genes is often detected because of lack of heterozygosity of chromosomal markers associated with deletions of segments of genetic material. Evidence for characteristic chromosomal abnormalities within tumor cells may lead to recognition of a tumor suppressor gene. Deletions of the tumor suppressor genes, p53 and the RB gene, have been detected in differentiated and undifferentiated thyroid cancer (136). Many chromosomal rearrangements are found in Hurthle cell tumors, and correlate with tumor recurrence (137).
Ret oncogene and Medullary Thyroid Cancer
Studies on patients with MENI and MEN II indicated linkage to chromosomes 11 (138) and 10, respectively. Subsequent studies demonstrated that the ret oncogene is present at 10q11.2. Germline mutations have been detected in this oncogene in all patients with MEN II and MEN III (or IIB), and familial MTC (139). RET is a cell-membrane receptor of the growth factor family, with tyrosine kinase function. In up to 97% of patients with MenIIA, mutations are found in codons 609,611,618, 620, and 630 in exons 10 and 11. These all involve substitutions of other aminoacids for cysteine, and are thought to cause activation of the gene by aberrant disulphide bonding causing dimerization. Similar changes are seen in Familial MTC. In patients with the MENIIB syndrome, almost all,if not all, mutations involve an amino acid substitution of threonine for methionine at codon (918) in exon 16, and are thought to induce a change in substrate phosphorylation. Somatic mutations in ret are present in up to half of patients with sporadic MTC and are almost always in codon 918 (140,141). Gene mutations in this codon are thought to imply a poor prognosis.
Familial tumors
Apparent familial thyroid cancer development has been reported by several clinicians, including cases which seem to show a dominant pattern of inheritance (142,143). Thyroid carcinomas occur rarely as part of several familial syndromes, which may involve hereditable loss of tumor suppressor genes. Papillary cancer can occur as an independent familial syndrome in 5-10% of patienst in different series. Whether the recurrence of PTC represents a true familial aggregation or rather the fortuitous association of PTC in the same family, is still a matter of discussion. However, recent evidence seem to support the existence of a true familial PTC syndrome based on the demonstration that familial PTC display the features of “genetic anticipation” (the disease recurs at an earlier age and at an higher aggressiveness in the 2nd generation compared to the first one) typical of familial diseases (144). In addition, a germline alteration consisting in short telomeres has been demonstrated in familial cases of PTC (145,146), which may be responsible for genomic instability leading eventually to thyroid cancer.
Other, more rare forms of familial thyroid tumors are those associated with complex hereditable diseases. Cowden’s disease is a familial syndrome which includes a variety of hamartomas, multinodular goiter, and carcinomas of several tissues including breast, colon, lung, and thyroid, especially in women (147). Thyroid carcinoma also co-occurs in patients with familial adenomatous polyposis of the colon (148), and can occur in the absence of bi-allelic inactivation of the APC gene. Differentiated thyroid carcinoma is reported to co-occur with chemodactomas of the carotid body, which can be inherited in a familial autosomal dominant form (149). Thyroid carcinoma is also associated with Gardner’s syndrome (150) and Carney’s Syndrome (151). Papillary thyroid carcinoma has been associated with papillary renal neoplasia in a distinct hereditable tumor syndrome. Some patients in the families also have nodular thyroid disease. The predisposing gene has been chromosome 1q21 (152). These syndromes are listed in table 18.5.
| Table 18.5. RARE SYNDROMES WITH HEREDITABLE THYROID TUMORS (NR9) | ||||||||||||||||||||||||||||||||||||||||
|
Experimental Thyroid Tumor Formation
Thyroid tumors have been induced experimentally in rodents by several procedures having as their common denominator a prolonged increase in pituitary thyrotropin production and thyroid stimulation. Goitrogenic drugs, if administered to animals for a prolonged period, can induce tumors, as numerous investigators (153) have demonstrated. These tumors are typically papillary adeno-carcinomas, and are associated with a diffuse hyperplasia of the thyroid gland. Old rats of some strains appear to develop thyroid cancers spontaneously
Roentgen irradiation of the thyroid and administration of 131I have both induced carcinomas in the experimental animal (154). A combination of 131I injury to the thyroid cell and prolonged administration of a goitrogen is especially likely to produce carcinomas, as shown by Doniach (155). Cell metabolism is altered by 131I, even when small amounts are administered. In the rat, 5 µCi prevents subsequent response to a goitrogenic drug (156). With larger doses the colloid is sparse, the follicles are variable in size, and large eosinophilic acinar cells appear. Very large doses of 131I (producing several thousand rads) to the rat thyroid radically alter cell metabolism, liberate TG within 1 or 2 weeks, and subsequently reduce the efficiency of hormone synthesis. 131I iodine irradiation in rats in doses so low as not to alter hormone biosynthesis immediately inhibits DNA synthesis and cell replication, as shown by a failure to respond to subsequent goitrogenic challenge. The cells also have a shortened life span. Similar inhibition of hyperplasia follows x-irradiation to the thyroid.
Therapeutic doses of 131I to patients also induce atypical nuclei, which may remain for many years (157). The doses of RAI needed to produce neoplastic change in the thyroid glands of animals closely parallel those given in the treatment of thyrotoxicosis in humans. The morphologic changes are intensified by a goitrogenic stimulus and reduced by thyroid hormone treatment.
The effects of radiation may be twofold. The nuclear morphologic changes may derive from an abnormality in cell division or replication of nucleic acids, which may predispose to carcinomatous change. Also, the damaged cell produces less thyroid hormone, and thereby ultimately comes under intense TSH stimulation, as in experiments with goitrogens. Thus, it seems certain that chronic TSH stimulation in animals is associated with the evolution of a neoplasm, especially if it is combined with radiation damage to the cell nuclei. Experimental thyroid tumors induced by 131I are initially TSH dependent. At first, they can be transplanted successfully only into thyroidectomized animals that are producing much TSH. After serial passages through several generations, the tumors may become autonomous and will then grow in a normal host. Partial or complete dependence on TSH is also observed in some human papillary and follicular tumors.
External Radiation and Thyroid Cancer
Duffy and Fitzgerald (158) first made the important observation that a high proportion of children with thyroid carcinoma had received therapeutic x-irradiation to the upper mediastinum or neck during childhood for control of benign lesions such as enlarged thymus, tonsils, or adenoids. Their finding has been amply confirmed (159-166). Winship and Rosvoll (167) studied 562 children with thyroid carcinoma from all parts of the world. Among those for whom adequate historical data were available, 80% had a history of prior x-ray treatment. This relationship is not so obvious for carcinomas developing after age 35. Significant x-irradiation to the head, neck, and chest in childhood increases the frequency of thyroid cancer by 100-fold (168), and the incidence is proportional to the dose, reaching at least 1.7% at 500 rads, or 5.5 cases per million exposed persons per rad each year (Fig. 18-6). Our own data dislose a 7% incidence by 30 years after irradiation (163). The latent period averages 10 -20 years, but tumors occur even after 20-40 years (Fig. 18-7). There appears to be no true threshold, since even doses as low as 9 rads increase the incidence of cancer (160). It is in fact probable that “natural” background radiation may produce many of the spontaneous tumors (169). There is a direct dose-response relationship through 1,000 rads (168). Higher doses of irradiation also induce tumors, and the true dose-response curve in the range 1,000 -5,000 rads in humans is not known. Benign nodules occur with nearly 10 times the frequency of cancers. Interestingly, the type of tumor induced is not different from those occurring spontaneously, and there is no relation between dose and latent period. For some reason, women are more prone to develop radiation-induced tumors than men, and both ethnic and familial factors may influence tumor development (170).
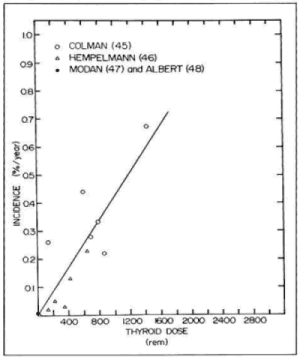
Figure 18-6. Estimated dose response for thyroid cancer in humans from external irradiation. The incidence of carcinomas each year is plotted against the original thyroid irradiation dose. (From Maxon H, Thomas SR, Saenger EL, Buncher ER, and Kereiakes JG. American J Med, 63:967, 1977)
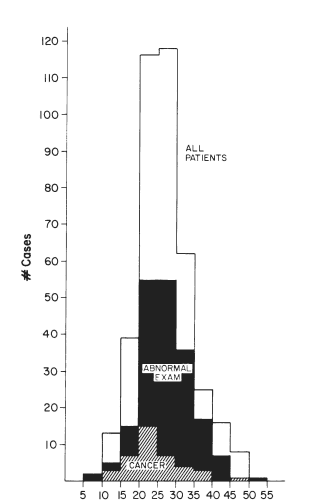
Figure 18-7. Distribution of patients with a history of irradiation to the head and neck, according to the time after irradiation at which they were examined. The majority of patients were seen 20 - 35 years after irradiation, but the incidence of tumors peaked 5 - 10 years earlier. Tumors continued to occur through 40 years after irradiation, and it is not clear that there is a finite latency period.
Probably any x-ray exposure of the thyroid has some carcinogenic potential, although the risk may decrease with age. Adults were extensively treated by x-irradiation for Graves’disease from 1930 to 1950. An increased incidence of carcinoma has been reported in these patients (171). A significant incidence of thyroid neoplasia was observed in patients who received x-ray therapy for thyroid disease (172). These patients were treated at ages up to 34, received 500-1,500 rads, and developed tumors 10-27 years after treatment. In a study of survivors of the atomic blasts at Nagasaki and Hiroshima, an increased incidence of thyroid cancer was found among persons who had received large amounts of radiation (173). Thus, the thyroid of the adult is sensitive to the carcinogenic action of x-rays, although not so sensitive as that of the child.
Radiation-associated tumors of the thyroid continue to occur, although x-ray treatment of thymic enlargement and tonsillar or adenoid hypertrophy has been discontinued since 1959. A recent analysis of 1787 patients treated with X-ray for Hodgkin’s disease found 1.7% to have thyroid cancer (174). The most dramatic and terrifying data emerged from the area around Chernobyl, where thousands of people of all ages received large doses of radiation from external fallout and ingested isotopes, especially short-lived isotopes of iodine. In this epidemic the risk of thyroid cancer is highest among children who were under 9 years and especially under 5 years old at the time of the Chernobyl explosion, and presumably ingested iodide via milk from cows grazing on contaminated forage. The latent period in these children is amazingly short (6 to 7 years), the tumors tend to be relatively aggressive, and are frequently associated with thyroid autoimmunity (175).
Radiation-associated tumors are generally found among younger patients. They are rarely undifferentiated, but some have been fatal. In a review of x-ray associated thyroid tumors at the University of Chicago Thyroid Clinic (163), the latent period among children treated predominantly in adolescence for tonsillar enlargement or acne averaged 20 years. It appears that the peak incidence of lesions is at 10 -25 years after exposure (Fig. 18-7, above), and it is probable that the occurrence of new cancers decreases over time. Among 100 consecutive patients seen in 1973 and 1974, only because they knew of prior radiation exposure, 15% had lesions suggestive of tumor and 7% had cancer proven at operation (162). Favus et al. (176) found a similar incidence of cancer (60/1056) in irradiated patients called back for evaluation. Although one case-controlled study suggests a lack of effect of radiation, the evidence, reviewed by Maxon et al (168), clearly confirms the importance of this problem.
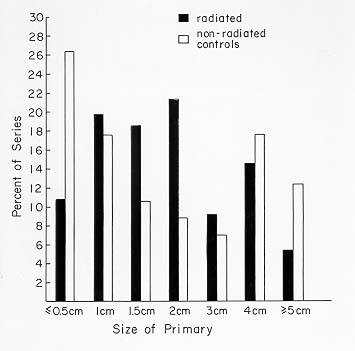
Fig 18-8. The size of radiation associated and non-radiation associated tumors was statistically non-different.
Based on these facts, it has been accepted by most physicians in the field that patients with a history of thyroid irradiation (over 20 rads, and certainly 50 rads) should be located and advised to have an assessment. This evaluation should consist at least of a physical examination and thyroid ultrasound. If one or more clear-cut nodules is found, then FNAC should be performed followed by surgical intervention in case of malignant result. Benign nodules are also found in these glands, with an incidence much higher than that of cancers. Serum TG levels tend to be elevated in irradiated patients, and antithyroid antibodies are more commonly present, but these tests are not of diagnostic value. When excised these glands often show multiple benign as well as malignant nodules as well as areas of fibrosis and hyperplasia (177).
M.P., 52-Year-Old-Woman: Thyroid Radiation and Multiple Gland Abnormalities
This patient was first seen with a history of irradiation for acne during her teens. She subsequently developed telangiectasia of the skin of her face. The month before the examination, she had observed a lump on the right side of the neck. Examination disclosed a 1-cm nodule in the right lobe of the thyroid and some irregularity of the left lobe. Thyroid scintiscan showed a cold nodule of the lower pole of the right lobe. Ultrasound examination of the right lobe identified a partially cystic nodule and a small cystic structure of the left. The FTI was slightly elevated at 10.9. RAIU was above normal. Thyroid antibodies were not present. Thyroid needle aspiration showed cells indicative of malignancy.
Routine blood tests disclosed alkaline phosphatase of 107 units (normal, 25-100 units), calcium 10.9 and 11.5 mg/dl (normal, 8.5-10.2 mg/dl), and phosphate 2.7 ng/dl. Repeated assay of FTI again demonstrated an elevated value of 15 (normal = 6-10.5). The level of parathyroid hormone was 0.65 ng/ml (with a coincident calcium level of 11.5 mg/dl), values indicative of primary hyperparathyroidism.
The patient was treated with potassium iodide for 1 week and admitted for ex¬ploratory surgery. A right upper para-thyroid adenoma weighing 908 mg was found. The adenoma showed areas of cystic degeneration and fibrosis. The thyroid gland was multinodular and was suspicious on frozen section for follicular carcinoma. There was extensive fibrosis around and adherent to the thyroid gland.A near-total thyroidectomy was performed. The gland weighed 17 g. Multiple nodules in the gland measured 1-18 mm in diameter. An 18-mm nodule in the right lobe was iden¬tified as follicular carcinoma. There were, in addition, multiple follicular adenomas and multiple Hurthle cell tumors, focal hyperplasia, and colloid nodules in the right and left lobe.
Postoperatively the patient received thyroid hormone. When seen 1 month after surgery, her calcium level was 9.4 mg/dl, phosphorus 3.5 mg/dl, and parathyroid hormone 0.28 ng/ml (normal). The FTI was 9.2 while taking 0.15 mg L-T4.
This patient developed a cystic parathyroid adenoma with hyperparathyroidism, multiple adenomas of the thyroid, follicular carcinoma, and multiple functioning adenomas that produced thyrotoxicosis. All of these tumors occurred concurrently in a gland showing changes typical of prior radiation exposure.
D.C., 19-Year-Old-Girl: Development of Thyroid Carcinoma, After X-Ray Therapy, While Receiving Thyroid Hormone
At age 10 the patient had respiratory distress and was found to have a superior-anterior mediastinal mass. There was left cervical lymphadenopathy and bilateral supraclavicular lymphadenopathy. Biopsy revealed Hodgkin’s disease of the nodular sclerosing variety. The result of staging laparotomy was negative. She was treated with 4,000 rads to a neck mantle field.
One year later the results of thyroid function tests were normal, but two years after x-ray treatment the FTI was 4.1 and the TSH level was 24 µU/ml. Thyroid hormone replacement therapy was begun, and the patient was carefully monitored over sub¬sequent years with periodic FTI and TSH determinations. Six years after irradiation a2-cm nodule was noted in the left lobe of the thyroid. This nodule was found to be cold on 123-I scan. Fine needle aspiration revealed cells suggestive of malignancy.
At surgery a papillary adenocarcinoma with capsular and vascular invasion was found, and a near-total thyroidectomy was performed. Postoperatively residual thy¬roid tissue was ablated by administration of 30mCi131I.The skeletal survey findings were negative; chest x-ray films and bone films were normal. She has remained free of evidence of thyroid carcinoma or Hodgkin’s disease in the subsequent three years.
This history demonstrates the occurrence of thyroid carcinoma, in a gland heavily irradiated during therapy for Hodgkin’s disease, while the patient was taking ade¬quate replacement doses of thyroid hormone. Possibly the period of X-ray induced hypothyroidism played a role in tumor induction. The tumor developed within six years of the radiation therapy. Fortunately, the tumor was not metastatic and has presumably been eradicated by surgery and RAI ablation of residual tissue.
In our series, postradiation carcinomas averaged 1.7 cm in size (Fig. 18-8), and 14% were below 0.5 cm.
The size distribution was similar to that of non-X-ray-associated tumors. They were more frequently multicentric than those in non irradiated glands and as aggressive, or more so, in behavior than tumors arising without known irradiation (178). The tumors are mainly papillary or follicular, but an occasional anaplastic cancer is also found.In examining patients, it should be remembered that benign and malignant salivary gland neoplasms, neuromas, parathyroid adenomas (179), laryngeal cancer, skin malignancies, and breast cancer also occur with undue frequency in this group of patients.
Thyroid nodularity and cancer also occur as a sequela of nuclear fallout. In the accident at Rongelap in the Marshall Islands (180), individuals received 200-1,400 rads. The incidence of nodularity was 40%, and nearly 6% proved to have cancer. Children in Utah exposed to small amounts of fallout from atomic bomb testing have been proven to develope nodules and possibly carcinomas.
Lack of Association of 131I Treatment and Thyroid Carcinoma
Iodine-131 treatment induces abnormalities in the thyroid gland that persist for many years (181). Giant nuclei, increased mitotic activity, hyperchromatic nuclei, and other abnormalities appear. It seems reasonable that these nuclear changes could lead to carcinomatous degeneration. Chromosomal damage in circulating lymphocytes has also been reported after 131I administration (182). Patients have developed thyroid nodules or tumors after 131I therapy for Graves’ disease; it has been suggested, but not proved, that the highest incidence has been among those treated during childhood. Some of the lesions found in 131I-treated children may actually have been carcinomas, but there has been debate (183) among the various pathologists who examined the specimens. Several reports of isolated instances of cancer after 131I treat¬ment of adults for Graves’ disease have appeared, but the large United States Public Health Service cooperative study failed to show an increased risk in this group (184-186). Studies by Holm et al (186) also failed to show an increase in cancer incidence among persons given 131-I either for diagnosis or for therapy for thyrotoxicosis. These patients were adults, and usually in the 40-60-year age group. Also, very large radiation doses may be less carcinogenic than small ones, and in half or more of these patients, the thyroid has been totally destroyed. Lastly, the follow-up time averages 8-13 years, which may be too soon to see radiation-induced neoplasia. Thus the evidence is reassuring but the question cannot be considered closed.
Thyroid Hyperplasia and Cancer
Chronic stimulation of the thyroid with TSH probably can lead to carcinogenesis in humans, as it can in animals. There are several reports of intensely hyperplastic congenital goiters, untreated for long periods, in which carcinomas have finally developed (187-191). Fortunately, most patients with congenital goiter are recognized and treated with replacement thyroid hormone at sometime during early childhood, so that chronic TSH stimulation does not occur. Interestingly, activating mutations of the TSH-R, which are metabolically like chronic TSH stimulation, lead to benign and not malignant change, as described above.
Relation of Cancer To Other Thyroid Disease
The relationship of thyroid tumors to other thyroid disease is still debated. In the preceding section we discussed whether carcinomas arise from adenomas, occurring either singly or as a component of a multinodular gland. While this must happen rarely, it is not the ordinary course of events. In support of this view one may note, for example, that, whereas adenomas are rarely if ever papillary, approximately 80% of all thyroid carcinomas are papillary. If carcinomas arise from adenomas, one might expect that the majority would be follicular rather than papillary, and this is not the case. Also, although carcinomas, largely of the papillary type, occur in nontoxic nodular goiters with a reported frequency of 4-17% of cases, the age of diagnosis of papillary carcinomas does not follow that for nontoxic goiter (192). Papillary carcinomas occur in children and adolescents, and reach their highest frequency during the middle decades of life. Multinodular goiter, by contrast, is infrequent in childhood, but increases with each decade. The high frequency of carcinomas detected in nodular goiter appears to reflect the efficiency of selection of patients for operation on the basis of suspicious clinical findings in the gland. Although it remains unproven, it is likely that in many or most thyroid adenomas and carcinomas, one specific mutational event leads directly to the development of the specific neoplasm.
Parathyroid adenomas occur in a small percentage of patients with thyroid cancer. The converse relationship may also exist; 2-11% of patients with parathyroid adenomas also have thyroid cancer (193-195). An important reason for this association is the induction of both tumors by X-ray exposure.
Neoplasia and Autoimmune thyroid diseases
An increased incidence of cancer in Hashimoto’s thyroiditis has been reported, Further, focal thyroiditis may occur as an immunologic response to thyroid cancer. In most series in the past the coexistence of Hashimoto’s thyroiditis among thyroid cancer patients was between 2-4%, but the real association, particularly with microcarcinomas, is difficult to be assessed because Hashimoto’s thyroiditis is rarely operated upon. Recently this issue has gained new attention in the era of FNAC, and several reports have found that when a thyroid nodule is associated with autoimmune thyroiditis, the chance of malignancy is significantly higher (9-40%) than in nodules not associated with thyroid autoimmunity (196,197). One possible explanation for this finding might be that patients with autoimmune thyroiditis tend to have higher levels of serum TSH (potential thyroid carcinogen) compared with non-autoimmune patients.
Many reports on Graves’ disease stress a normal or low coincidence of cancer, but several series have reported a significant association between Graves’ disease and thyroid cancer, ranging from 3 to 10% (198-200). However, most of these series were surgical, and the patients were selected for surgery on the basis of suspicious nodules or large goiters. In Graves’ patients treated by radioiodine, no subsequent increase in the discovery of thyroid cancer has been reported.In our review, 4 of 50 patients with thyroid cancer had coincident Graves’ disease (201). Belfiore et al found the risk of thyroid cancer in Graves’ disease to be increased 2-3 fold (198). TSAb can stimulate thyroid cancers when Graves disease coexists, so the idea that TSAb might induce malignant change is tenable, but not proven. It also is possible, but unproven, that continued stimulation of a tumor may make it behave in a more aggressive manner (198,202). Patients with Graves disease and thyroid cancer who underwent total thyroidectomy and 131I ablation fared as well in follow-up as did patients without Graves’ disease.
Micro-carcinomas
The term micro-carcinoma refers to tiny carcinomas (<1 cm), usually papillary and often sclerotic. Such tumors were frequently found in autopsy studies when the thyroids were sectioned completely at 1-2 mm intervals and every abnormality was studied, or during histology of glands operated for large multinodular goiters (203,204). Nowadays, with the extensive use of thyroid ultrasound, micronodules (<1 cm) are frequently discovered in the general population, and proven malignant in a significant percentage. These tumors are probably synonymous with the "occult" tumors described at autopsy, but since they are discovered during life can no longer be considered occult. The term microcarcinomas is now preferred to occult.
Tiny carcinomas, usually papillary and often sclerotic, have been found in 5.7% of thyroids of adults coming to autopsy in the United States (203). This prevalence is noted only when the thyroid is sectioned completely at 1-2 mm intervals and every abnormality is studied. The tumors have a mean diameter of 2 mm, and almost all are under 5 mm. The prevalence is best known in adults (204) and may be lower in young people. Since, in some glands, collections of psammoma bodies also exist in tiny scarred areas, it is hypothesized that such lesions may spontaneously regress. Because of their small size, they are detectable only at surgical or pathologic examination of the gland. These observations, now widely accepted, have provoked much discussion. Certainly most of these tiny tumors cannot be biologically significant, considering the low incidence of clinically recognized cancer. Most of the lesions are probably missed during routine surgical or autopsy pathologic studies. Their cause is unknown. They may be a variant of the occult sclerosing carcinomas described by Hazard, but the latter tumors are usually larger, have a predominant sclerotic component, and clearly do metastasize. It has been suggested that such lesions are in fact the cancers found after thyroid irradiation, but most likely they can explain only a small proportion of radiation-associated tumors. Most of these lesions would go undetected in a standard surgical pathologic examination, and the great majority of radiation-associated lesions are larger. In our own series, the radiation-associated le¬sions were on average 1.7 cm in diameter and only 14% were under 0.5 cm. Whether such "minimal" cancers are in fact the occasional precursors of clinically evident cancers is a moot point. It is clear that they are at present an important pathologic -but not clinical --entity.
PATHOLOGY (Figures 18-9a-h)
Pathologists are agreed that there are peculiar difficulties in the classification and diagnosis of malignant tumors of the thyroid. The histologic changes required for diagnosis of carcinoma include absence of a true capsule, invasion of surrounding normal tissue, invasion of blood and lymph channels, loss of normal follicular architectural arrangements, and cellular abnormalities such as an increase in the ratio of nucleus to cytoplasm, enlarged vesicular nuclei, nuclear folding, increased mitoses, and hyperchromasia of the nucleus. Recently, aneuploidy of nuclear DNA content has been added to this list. Obviously the presence of distant metastases is the most certain criterion. Most students of the disease agree that the ordinary criteria of malignancy have little prognostic value in thyroid tumors, except perhaps in the wildly growing anaplastic tumors. However, it may be noted that pathologists at the Mayo Clinic believe a histologic typing by their criteria provides significant information on prognosis.
Examples of the histologic patterns of several of these tumors are given in Figure 18-9. The papillary adenocarcinoma typically shows tumor cells around a fibrovascular core and, not infrequently, areas of follicular differentiation. Papillary lesions tend to be infiltrative, and encapsulation is rare. Lymphocytic "reactions" are prominent. The cell nuclei have a ground-glass or "cat’s eye" appearance and intracellular inclu¬sions are common. Vascular invasion is rare. Psammoma bodies are often abundant. Multiple intraglandular foci are frequent, especially in children. Areas of lymphocyte infiltration, and even extensive lymphocytic thyroiditis, are common, especially in papillary tumors. Many tumors look much like follicular cancers, but have the characteristic nuclei of papillary cancers. These constitute the "follicular variant" of papillary cancer, and behave more or less as do other papillary cancers.
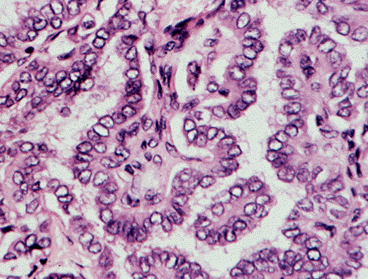
Figure 18-9. A) Papillary carcinoma of the thyroid. The structure is made up of complex (branches on branches) fibrovascular core structures covered by crowded, overlapping, vesicular nuclei (artifact of fixation). Little colloid is visible. Such histologic foci may be encapsulated, sclerosing, invasive, or multicentric.
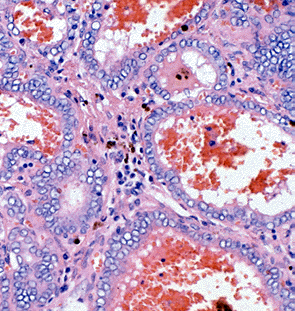
Figure 18-9 B) Follicular variant of papillary carcinoma with more typical vesicular nuclei, and hemorrhage in follicular lumens.
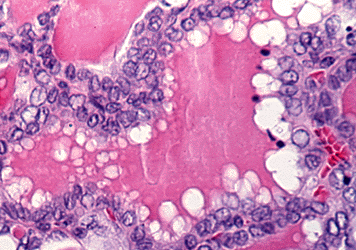
Figure 18-9. C) Follicular variant of papillary carcinoma with crowded nuclei showing nuclear folding and peripherally vacuolated colloid.
Follicular adenocarcinomas vary from those with a definite follicular pattern to those with solid sheets of cells. The lesions are more frequently encapsulated, but capsular and blood vessel invasions are typical. The nuclei are normo-or hyper-chromated, or may be quite vesicular. One variant, the so-called malignant adenoma, appears to be nearly benign and can be identified as malignant only by the demonstration of invasion of vessels or capsule, or because of the presence of distant metastases, which may also be composed of normal-appearing thyroid tissue. Hurthle cell carcinomas usually grow as solid sheets of large eosinophilic granular cells with much cytoplasm, and less often with a follicular pattern. An Hurthle cell appearance can also be observed in some papillary tumors. (Figure 18-9d)
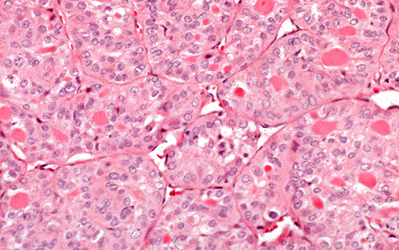
Figure 18-9. D) Poorly differentiated follicular carcinoma with oxyphillic features.
Medullary tumors (Fig. 18.9 f-g) have an ominous histologic pattern, with solid masses of cells with large vesicular nuclei (205). There may be considerable associated fibrosis, and deposits of amyloid are a helpful diagnostic point. At the time of initial histologic examination the pathologist should recognize these tumors as entities distinct from the undifferentiated cancers, for the medullary carcinomas have a much better prognosis. Medullary thyroid carcinoma (MTC) is associated with amyloid deposition in the surrounding tissues. Recent studies demonstrated that full-length calcitonin is the sole constituent of amyloid in MTC (206).
In the undifferentiated group of small-cell tumors, giant-cell tumors, and carcinosarcomas, or in the miscellaneous group, the histologic pattern has little resemblance to the original thyroid structure (Fig 18-9e-h).
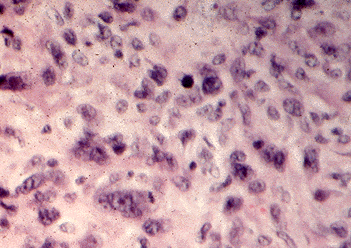
Figure 18-9. E) An anaplastic carcinoma of the thyroid with pleomorphic giant tumor cell nuclei.
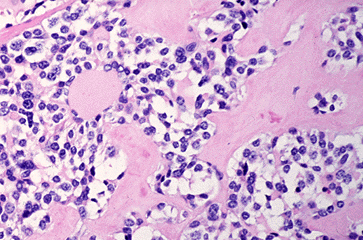
Figure 18-9. F) Medullary (C-cell) carcinoma of the thyroid with amyloid stroma.
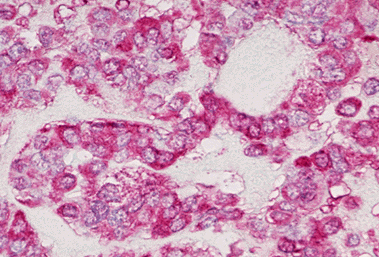
Figure 18-9. G) Immunohistochemical anti-calcitonin antibody stain of a medullary carcinoma showing strong red positivity.
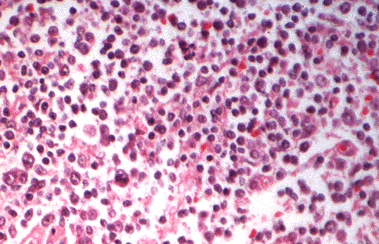
Figure 18-9. H) Large cell lymphoma of the thyroid.
The general experience of pathologists has been that, in the absence of irradiation, the substrate in which thyroid tumor forms is usually normal thyroid tissue or displays the changes of multinodular goiter or adenoma in approximately the proportion found in any sampling of the general population (207). There is a slightly increased frequency of association with benign adenomas and with Hashimoto’s thyroiditis (208). Lymphomas are associated with Hashimoto’s thyroiditis, and there is considerable evidence that lymphoma actually evolves from a gland with thyroiditis (209).
Multicentricity is a common feature of thyroid cancer, especially papillary cancer. Innumerable separate foci are sometimes found. Estimates of multicentricity range from 20 to 80% (210,211). Whether this phenomenon represents truly multicentric sites of origin or intrathyroidal dissemination is not clear. This multifocality is thought to be one cause of recurrences in patients treated by subtotal rather than total thyroidectomy.
Both papillary and follicular tumors may appear as small (less than 1.5-cm) tumors surrounded by a densely fibrotic reaction. Although it is frequently said that these "occult" (because they may be found incidentally at operation) tumors are benign, the original report by Hazard (212) and subsequent studies show that cervical lymph node metastases occur (213).
Occasionally pathologic examination suggests conversion of differentiated papillary or follicular cancers into anaplastic forms or conversion of a follicular adenoma into a follicular carcinoma.
An interesting aspect of thyroid tumor pathology is the frequency of metastatic tumors to the thyroid, 5% in the data of Silverberg and Vidone (214) for unselected autopsies and 24% for patients dying of metastatic malignant disease.
Course of the Disease
Clinical signs
Most frequently the tumor is discovered accidentally by the patient or physician as a lump in the neck or may be a fortuitous finding at ultrasound of the neck. Less and less frequently, nowadays, it may appear as a gradually enlarging, painful mass with associated symptoms of hoarseness, dysphagia or dysphonia, or there may be difficulty in breathing. Occasionally a arrives with metastatic nodules in the neck, pulmonary symptoms from metastases, or a pathologic fracture of the spine or hip. Usually there are no symptoms of hyper-or hypothyroidism, but in very rare instances the tumor, usually metastatic follicular carcinoma, can produce enough hormone to cause hyperthyroidism (215,216). (See also Chapter 13)
Upon examination of the neck, carcinoma of the thyroid characteristically appears as an asymmetrical lump in the gland. If it is still within the confines of the gland, it will move with the gland when the patient swallows and may be moveable within the gland. If it has invaded the trachea or neighboring structures, it may be fixed; this is a useful sign. Lymph nodes containing metastases may be found in the supr-aclavicular triangles, in the carotid chain, along the thyroid isthmus, and rarely in the axillary nodes. A sentinel or "Delphic" node above the isthmus may be present. Although carcinoma of the thyroid is typically firm or hard, rapidly growing lesions may sometimes be soft or even fluctuant. When the tumor is poorly differentiated or anaplastic, the lesions may undergo necrosis and discharge through sinuses that developed in the skin of the neck. Patients with thyroid cancer are also prone to develop other cancers, the risk being about double the average. Among these cancers is an excess incidence of leukemia, perhaps related to 131I therapy (217-219).
Age at diagnosis has an important bearing on the patient’s subsequent course. The adverse effect of age on the prognosis increases gradually with each decade (220). For practical assessment purposes, it is clear that patients diagnosed before age 45 have a much better prognosis than those detected later (221). Age is also directly related to the incidence of undifferentiated tumors and to overall mortality. Pregnancy does not seem to worsen the course of established or previously treated thyroid can¬cer (222). Overall, women have a better prognosis than men with cancer (223). Other characteristics of the tumor, including (as would be expected) extraglandular extension, gross invasion of the tumor capsule, and increasing size also carry a worsened prognosis (223).
Papillary Carcinoma-course of disease
Papillary carcinoma has a peak incidence in the third and fourth decades (224). It occurs three times more frequently in women than in men, and accounts for 60-70% of all thyroid cancers in adults and about 70% of those found in children. The disease tends to remain localized in the thyroid gland and in time metastasizes locally to the cervical or upper mediastinal nodes. The lesions are multicentric in 20% or more of patients, especially in children. Using rigid pathologic criteria, perhaps two-thirds of predominantly papillary thyroid cancers are found to have follicular elements. The natural history of these tumors is similar to that of pure papillary lesions (225). The metastases may conform to either histologic pattern. At present, the mixed tumors are lumped together with all other papillary cancers. This tumor tends to be indolent and may exist for decades without killing the host. In a Mayo Clinic series of papillary tumors that were detected because of lymph node metastasis or found incidentally during surgery of the thyroid gland, all the patients were unaffected by the tumors over several decades (224). However, in a recent series from one single institution in Italy, it is apparent that the way of presentation of papillary thyroid cancer has been changing in the last to decades compared to previous years. In particular the authors report increasing number of small tumors and less frequent lymph node metastases at presentation (226)
The frequent occurrence of occult or “minimal’ incidentally found thyroid cancers, usually papillary and under 0.5 cm in size, is described above. However, the term occult has been used in a variety of ways, including reference to tumors with malignant nodes but no obvious primary, or in reference to any tumor under 1.5 cm in diameter. Mayo Clinic reports of papillary tumors under 1.5 cm in diameter, treated with conservative subtotal thyroidectomy and node dissection, have stressed their non-lethal nature, but a 1980 follow-up report on 820 patients treated by this group notes that 6 patients eventually died after spread of tumor from such "occult" primaries (227). Patients with appropriately treated Clinical Class I or II lesions have 96-100% survival even after 15-30 years. Survival lowers to 87% for Class III and 35% for Class IV lesions at 15 years.
While the disease may be aggressive in children, it is distinctly less aggressive in young adults, as compared to patients over age 40 (223).Young patients tend to have small primary lesions and extensive adenopathy, but even with local invasion survival is good (228). When papillary cancer occurs in persons over the age of 45, it may show, on microscopic examination, areas of undifferentiation, and pursue a highly malignant clinical course. The lesions tend to be larger and more infiltrative, and to have fewer local metastases (229). It is possible that persons dying in older age actually have had their disease since youth, and that it has simply evolved into a more malignant phase (230,231).
Papillary carcinoma tends to metastasize locally to lymph nodes, and occasionally produces cystic structures near the thyroid that are difficult to diagnose because of the paucity of malignant tissue. In this case measurement of thyroglobulin in the fluid aspirate is the clue for the correct diagnosis. The presence of nodal metastasis correlates with recurrence (230-232) but has little effect on mortality in patients under age 45. In old studies, cervical adenopathy even seems to confer a protective effect on young people, but this assumption is not confirmed in present series. Indeed in patients over 45, the presence of nodes is associated with greater recurrence rates and more deaths (233,234). The tumors often metastasize elsewhere, especially to lung or bones.
Papillary tumors may metastasize to the lungs and produce a few nodules, or the lung fields may have a snowflake appearance throughout. These tumors are amazingly well tolerated and may allow relatively normal physical activity for 10-30 years. At times, particularly in the follicular variant of papillary thyroid cancer, the pulmonary metastases are active in forming thyroid hormone, and may even function as the sole source of hormone supply after thyroidectomy. The metastases may progress gradually to obstructive and restrictive pulmonary disease. They also may develop arteriovenous shunts, with hypoxia or cyanosis. Such shunts become more prominent during pregnancy, perhaps as an effect of the increased supply of estrogens.
The tall cell variant of papillary carcinoma comprises about 10% of total cases, and as noted by several authors appears to be more aggressive than other forms of the disease (235,236). .
The usual net extra mortality in papillary cancer is not great when compared to that of a control population, perhaps 10-20% over 20-30 years (231,232,234). Mortality is rare in patients diagnosed before age 40, and is much greater in the patients found to be in clinical stages III and IV at initial diagnosis. About one-half of patients ultimately dying from this lesion do so because of local invasion. Frazell and Duffy (237) have noted that papillary carcinoma is not always so benign; they reported 35 patients with "invasive papillary carcinoma," which had a very malignant course.
We found that risk of death from cancer was increased by extrathyroidal invasion (6 fold) or metastasis (47 fold), age over 45 years (32 fold) and size over 3 cm (6 fold). Thyroiditis, multifocality and the presence of neck nodes had no effect on disease-induced mortality.
P.P. 41-Year-Old Man: Long-Term Survival with Papillary Cancer
This man was first seen at age 41. At age 14, while living in Yugoslavia, he had developed a mass in his neck. Tracheostomy was required because of dyspnea, and a biopsy of the mass was performed. A diagnosis of papillary thyroid carcinoma was made, and he was treated with radiotherapy to the neck. Because of an abnormal chest x-ray film, presumed to be due to tuberculosis, he was given streptomycin and isoniazid for 2 months, but this therapy was discontinued when no improvement occurred. Extracts of calf thymus were injected. He was evaluated for hemoptysis at age 18, and chest x-ray films again showed the infiltrates without evidence of change. The patient was not given thyroid hormone replacement therapy at any time. At age 39 he developed fatigue, substernal chest pain, occasional cough without production of sputum, and occasional hemoptysis. A thyroid scintiscan at another hospital revealed poor visualization of the thyroid and uptake of 4%, and 131I uptake in the mediastinum.
Physical examination disclosed hyperpigmentation of the skin in the area of radiotherapy in the neck and supraclavicular areas. There was no evidence of a mass or lymph-adenopathy. The tracheostomy site was well healed. The lungs were clear, BP was 100/70, pulse 68, and respiration 16/min, and there was no cyanosis. Rou¬tine complete blood count, urinalysis, and blood chemistry test results were normal. Chest x-ray films showed multiple nodular densities throughout both lungs and a prominent left hilum. The results of a radiographic skeletal survey and a technetium pyrophosphate bone scan were normal. The FTI was 8.4 and TSH was 4.1 µU/ml. A whole body 131I scan showed multiple areas of abnormal uptake in the area of the thyroid, and satellite areas of focal uptake around the thyroid bed. There was also focal activity in the mediastinum, in both hila and in the lung fields. A 72-hour chest uptake was 8.9%. The serum TG level was 81 ng/ml.
The patient was treated multiple times with RAI and remained well while taking replacement T4 for ten years. At age 51, the tumor grew more rapidly, failed to accumulate 131I, and caused the patient’s death.
This patient developed thyroid carcinoma at age 14 and probably had lymph node and lung metastases at that time. He lived a normal life during the ensuing 27 years, without suppressive thyroxine treatment, and with only intermittent episodes of hemoptysis. The tumor responded at first to 131I but later was uncontrolled. The common benign course of metastatic papillary thyroid carcinoma over many years is clearly shown, as is the equally typical later exacerbation and death.
Follicular Carcinoma
Follicular carcinoma has a peak incidence in the fifth decade of life in the United States and accounts for about one-quarter of all thyroid carcinomas (223,238,239). In past decades, follicular carcinomas comprised up to 50% of thyroid malignancies in Europe. The high incidence may partly be explained by iodine deficiency, but, more likely, was due to histological miss-classification at a time when the follicular variant of papillary thyroid carcinoma was not recognized as papillary but rather was classified as follicular. It is a slowly growing tumor and frequently is recognized as a nodule in the thyroid gland before metastases appear. Variation in the cellular pattern ranges from an almost normal-appearing structure to anaplastic tissue that forms no follicles or colloid. The insular variant of follicular tumor tends to be more aggressive (240). The tumor is three times as common in women as in men. At operation one-half to two-thirds of these tumors are resectable. Tumors that are small and well circumscribed (not surprisingly) tend to be less lethal than those actively infiltrating local structures at the initial operation. Local adenopathy, which is uncommon, probably carries a greater risk, and extensive invasion of the tumor capsule and thyroid tissue increases mortality (241). Local direct invasion of strap muscles and trachea is characteristic of the more aggressive tumors (242). Resectability depends on this feature, and death may be caused by local invasion and airway obstruction. The "minimally invasive" variant has a far better prognosis than the highly invasive variant.
Follicular carcinomas tend to invade locally and metastasize distantly, rather than to local nodes, and are especially prone to metastasize to bone or lung. In a Massachusetts General Hospital series (231), one-half had metastasized at the time the diagnosis was originally established. Bony metastases are usually osteolytic, rarely osteoblastic, and the alkaline phosphatase level is rarely elevated. The tumor and metastases often retain an ability to accumulate and hold iodide, and are therefore sometimes susceptible to treatment with RAI. Indeed, some metastatic tumors synthesize thyroid hormone in normal or even excessive amounts. RAI therapy, as discussed below, improves survival in these patients (241).
Occasionally the primary lesion of a follicular tumor appears to be entirely benign, but distant metastases are found. Invasion of vessels or the capsule, apart from the metastasis, is the only reliable criterion of malignancy. This variant has been called the “benign metastasizing struma” or malignant adenoma. It has a more prolonged course than do other varieties of follicular tumor, and is the type that has offered the best opportunity for the therapeutic use of 131I.
The net extra mortality attributable to follicular cancer in the 10 -15 years after diagnosis is 30 -50% (231,233,235). Of the patients dying from the lesion, three-fourths do so from the effect of distant metastases and the remainder from locally invasive disease.
Hurthle CellTumors
Hurthle cell tumors are histologically distinct from other follicular tumors, but they pursue a similar course. They tend to invade and metastasize locally and have a strong propensity to recur after surgery. The course tends to be prolonged. These carcinomas often do not accumulate 131I. However, in a large survey, Caplan et al (243) found that 4.4% of Hurthle cell neoplasms were hot on scan and 8.9% were warm. Serum TG levels may be normal or elevated. Cheung et al recently studied the presence of ret/PTC gene rearrangements in Hurthle cell tumors and found that many expressed ret/PTC, and also had other evidence of a papillary cancer origin, including focal nuclear hypochromasia, grooves, and nuclear inclusions. Tumors with the ret/PTC gene rearrangement tended to have lymph node metastases, rather than hematogenous spread. Thus Hurthle cell tumors can be classified into Hurthle cell adenomas, Hurthle cell carcinomas, and Hurthle cell papillary thyroid carcinoma (244).
Insular Tumors
A subset of thyroid carcinomas which give a histologic picture of islands of cells -thus "insular" --has been identified (243). These tumors often look like anaplastic cancers, but sometimes are able to concentrate 131I and thus are amenable to this excellent treatment. Whether these are properly considered a variety of follicular cancer is uncertain. The important message is that the histology in this instance does not reliably predict the utility of 131I treatment, suggesting that all patients with thyroid cancer should at some point be studied to determine whether 131I treatment is possible.
Undifferentiated Tumors
Undifferentiated tumors occur predominantly in persons over 50 years of age and constitute an increasing proportion of lesions in each subsequent age decade. Of great interest is the pathologic evidence that such tumors arise, in perhaps half of the cases, in a long-standing benign lesion or in differentiated carcinoma (245). Although 131I therapy for differentiated cancers has been blamed for this dedifferentiation, current evidence is against this hypothesis. Spindle cell, and most giant-cell carcinomas of the thyroid grow rapidly and are very invasive. Local invasion may cause difficulty in breathing or swallowing, and tracheotomy is frequently required. These tumors metastasize to lymph nodes both locally and widely, but not characteristically to bone. Pulmonary metastases are frequent. Some patients present with a tender mass suggesting thyroiditis, and occasionally thyroid destruction induces hyperthyroidism (246). The outlook in this particular group of tumors is poor. By the time the diagnosis is made, the disease has spread in most patients beyond the area that can be attacked surgically, and they die within 6 months to 1 year. A few, perhaps 10%, of these tumors are entirely resectable when first discovered. There is nothing characteristic about the growth pattern of these tumors; their behavior is similar to that of any highly malignant tumor elsewhere in the body. The course of the epidermoid carcinoma and sarcoma of the thyroid is essentially the same. Small-cell anaplastic carcinomas are also found, but probably most tumors so classified in the past are actually lymphomas or lymphosarcomas.
Malignant Lymphomas
Lymphomas of the thyroid gland represent less than 5% of primary thyroid neo¬plasms (247-250). Unlike most other thyroid neoplasms, lymphomas usually appear as rapidly enlarging masses and local symptoms are common. Many patients note pain, hoarseness, dysphagia, and dyspnea or stridor. Hoarseness is often present in the absence of vocal cord paralysis. Rarely, patients may have the superior vena cava syndrome. The mean age at occurrence is 62 years. Primary lymphomas of the thy-roid are two to three times more common in women than in men.
The incidence of hypothyroidism at the time of appearance is variable, ranging from 0 to 60% (251,252). The co-occurrence of pathologic lymphocytic thyroiditis has ranged from 30 to 87%. These figures may underestimate the true incidence, as some patients have only had a biopsy examination and in others the entire gland has been replaced by the lymphoma. The frequent presence of thyroiditis has naturally led to the suggestion that the lymphoma might derive from preexisting thyroiditis.
The co-occurrence of thyroiditis may create difficulties in the proper interpretation of fine needle aspiration cytology. The clinical appearance must be carefully considered in accepting a diagnosis by fine needle aspiration of thyroiditis only or thyroiditis with lymphoma. An excisional or large needle biopsy may be necessary to make the correct diagnosis.
The majority of thyroid lymphomas are diffuse, large-cell lymphomas (formerly classified as diffuse histiocytic or reticulum cell lymphomas), diffuse mixed small and large cell lymphomas (formerly called diffuse mixed lymphocytic-histiocytic), or diffuse small cleaved-cell lymphomas (formerly classified as diffuse poorly differen¬tiated lymphocytic). Although older series include reports of Burkitt lymphoma, none were reported in larger, more recent series. Areas of diffuse large-cell lymphomas may have features similar to those characteristic of Burkitt’s lymphoma or the Reed-Sternberg cells of Hodgkin’s lymphoma.
Metastatic Carcinomas to the Thyroid
Melanomas, breast tumors, pulmonary tumors, gastric, pancreatic, and intestinal carcinomas, renal carcinomas, lymphomas, carcinomas of the cervix, and tumors of the head and neck may metastasize to the thyroid. Sometimes the first indication of one of these tumors may be the appearance of a lump in the neck. Unless there is evi¬dence for a primary site elsewhere, these tumors are easily mistaken for expanding tumors that have their origin in the thyroid gland. Usually by the time metastases appear in the thyroid, other metastases have occurred and the primary lesion may be discerned.
Cancer in Aberrant Thyroid Tissue
Thyroid tumors occasionally arise in lingual thyroids, along the thyroglossal duct (253), in substernal goiters, and even in struma ovarii. Whenever discovered, the therapeutic approach should the removal of the tumor assoiated with total thyroidectomy to allow subsequent treatment with radioiodine and follow-up based on serum Tg mreasurement.
MEDULLARY CARCINOMA
Hazard et al. (205) first described a unique tumor in the thyroid characterized by sheets of cells with large nuclei, amyloid deposits, fibrosis, multicentricity, and an unexpectedly benign course in view of the solid tumor appearance. Over 50% may have local or distant metastases at diagnosis. The tumors may metastasize locally, or to bones and soft tissues. The thyroid primary tumor and the metastases may show dense calcification on x-ray film. The course tends to be progressive, and 10-year survivorship varies from 50 to 70%. These tumors, which constitute 2-8% of all thyroid cancers, are derived from the "light," or "C," or "parafollicular" cells. These are calcitonin (CT)-secreting cells, distinct from thyroid acinar cells, and are of ultimo-branchial origin.
The tumors may occur sporadically (about 70% of the total) or as part of the MENII syndromes, which constitute about 10-20% of the cases, and are transmitted in families as dominant traits due to activating germline point mutations of the RET proto-oncogene (254,255). In contrast with thyroid epithelial cell tumors, the female to male ratio is near unity. MEN-II (or IIA) includes patients with medullary thyroid cancers, pheochromocytomas, and parathyroid hyperplasia or adenomas. MEN-III (or MEN-IIB) includes medullary thyroid carcinoma, mucosal neuromas, pheochromocytomas, which are usually bilateral and often malignant, occasionally cafe-au-lait spots, and possibly Gardner’s syndrome (mucocutaneous pigmented nevi and small intestinal polyps) (133,256-259). An occasional variant of medullary thyroid cancer appears to contain both CT and TG, suggesting the cells, surprisingly, have features of both medullary thyroid cancer and follicular cancer (260).
Gastrointestinal symptoms including diarrhea, constipation, and rarely megacolon occur in these patients and may occur before the thyroid tumor is detected. Hyperplasia of C cells often precedes the development of familial cancers (261). Medullary tumors derived from the C cells not only secrete CT, but in addition may produce serotonin (with a carcinoid syndrome), prostaglandins, corticotropin releasing factor, and adrenocorticotropic hormone (causing Cushing’s syndrome), histaminase, and somatostatin. Some of these substances, rather than calcitonin, may be the cause of the severe diarrea syndrome associated with metastatic medullary thyroid cancer. Interestingly, expression of somatostatin appears to correlate with improved prognosis (262). Medullary thyroid carcinoma cells in tissue culture have been found to produce Ghrelin, which is an endogenous ligand for the GH secretagogue receptor. MTC is known to produce several gastrointestinal hormones and neuroendocrine peptides, in addition to calcitonin, and including CGRP, ACTH, serotonin, chromogranin A, and vasoactive intestinal peptide, and to this we now add Ghrelin (263). Alcohol ingestion is reported to induce attacks of flushing and diarrhea and to stimulate CT secretion by the tumors (264). The CT secreted by these tumors rarely causes hypocalcemia (265).
Medullary tumors have an ominous histologic pattern, with solid masses of cells with large vesicular nuclei (212). There may be considerable associated fibrosis, and deposits of amyloid are a helpful diagnostic point. At the time of initial histologic examination the pathologist should recognize these tumors as entities distinct from the undifferentiated cancers, for the medullary carcinomas have a much better progno¬sis.
Ret oncogene and MedullaryThyroid Cancer
Studies on patients with MENI and MEN II indicated linkage to chromosomes 11 and 10, respectively (138). Subsequent studies demonstrated that the ret oncogene is present at 10q11.2. Germline mutations have been detected in this oncogene in all patients with MEN II A and MEN IIB, and familial MTC (139). RET is a cell-membrane receptor of the growth factor family, with tyrosine kinase function. In up to 97% of patients with MenII A, mutations are found mostly in codons 609, 611, 618, 620, and 630 in exons 10 and 11. These all involve substitutions of other aminoacids for cysteine, and are thought to cause activation of the gene by aberrant disulphide bonding causing dimerization. Similar changes are seen in Familial MTC. In patients with the MENIIB syndrome, almost all, if not all, mutations involve an amino acid substitution of threonine for methionine at codon 918 in exon 16, and are thought to induce a change in substrate phosphorylation. Somatic mutations in ret are present in up to half of patients with sporadic MTC and are almost always in codon 918 (140,141). Mutations in this codon are thought to imply a poor prognosis (266).
Calcitonin and CEA
The calcitonin assay provides a convenient screening procedure in families with this genetic trait (267). Family members at risk should firstly BE screened for ret oncogene mutation in their blood, and when when recognized as a carrier of the mutation, should be screened by neck ultrasound and calcitonin measurement to assess the presence of the disease. Every member of one of these families with either a thyroid mass or elevated calcitonin levels should have a thyroidectomy. If no thyroid nodule is detected and the serum calcitonin is normal according to the reference range for that specific age group, prophylactic thyroidectomy should be considered in order to take out the thyroid gland before the disease is initiated.
There is controversy regarding the more appropriate age for prophylactic thyroidectomy. Recent ATA guidelines for MTC have suggested different time for surgery according the the age and the mutation found (268) In MEN-IIA, the tumors follow a rather benign course somewhat akin to that of follicular cancer, and usually can be controlled by surgery. MEN-IIB tumors are much more aggressive and often cause death in the second or third decade. TSH suppressive treatment is not efficacious in MTC and has no logical basis.
Secretion of calcitonin by medullary cancer is remarkably increased by calcium or pentagastrin infusion (269). This procedure can be helpful in establishing a diagnosis if available. At present the infusion of pentagastrin (0.5 µg/kg over 5 seconds), with determination of calcitonin levels at 0, 1, 2, 5, 10, and 15 minutes, appears to be the best test. Alternatively stimulation with calcium gluconate, 20 mg/kg, can be done with infusion IV over 1 minute and testing as tor TRH. (This test can induce adverse cardiac effects, and close monitoring is reasonable.) Basal CT values are normally under (depending on the laboratory) 30 pg/ml (269). Values of 30-100 after pentagastrin indicate hyperplasia, and values over 100 typically indicate the presence of cancer. Calcitonin should drop to undetectable levels if the tumor is completely removed surgically. It should be noted that excess production of CT is not unique for medullary cancer, but can occur with granulomatous diseases and other cancers. An alternative is to monitor Calcitonin after Calcium infusion. Patients with the syndrome should also be studied with parathyroid hormone and catecholamine assays in order to determine the presence of other components of the syndrome.
The neoplastic C cells also produce carcinoembryonic antigen (CEA) in large amounts. Serum CEA levels are elevated in medullary cancer with the same frequency as are CT levels (270). Although CEA determination provides another parameter to follow, it does not offer any obvious advantage, and lacks the specificity of CT determinations. Tumor dedifferentiation is associated with a fall of CT and increasing CEA. This is an ominous sign.
Several specialized scanning procedures have been used in MTC. Total body imaging with Tl-201 chloride andTc-99m(V)DMSA have been successful in localizing metastases. 131I MIBG and 131I anti-CEA have been used both for localization and in attempts at therapy (271). Most recently, radiolabelled somatostatin has been used as a whole body scanning agent.
Since patients with MENIIA and IIB, and familial MTC, probably all have a germline mutation of the ret oncogene near the centromere of chromosome 10, and many patients with sporadic MTC have somatic ret mutations, molecular techniques can be used to detect at-risk subjects in recognized families (269). PCR amplification of exons 10, 11, and 16, followed by single strand conformational electrophoretic analysis, or use of specifically designed restriction enzyme sites, allows recognition of most mutations (272). This information is crucial in defining potential risk in young children and identifying need for operation or frequent pentagastrin testing (255), and presumably will also screen out members of families who will not need to be repetitively screened by pentagastrin stimulation tests. Unlike occasional false positives with CT assays, the results of genetic screening are unambiguous (273).
For readers with an interest in a somewhat broader view of oncogenesis, it may be noted that a syndrome entirely analogous to metastatic medullary thyroid carcinoma appears frequently in aged bulls (274). Histologically similar adenomas are also frequently found.The lesion may be due to excessive dietary calcium; whether a similar stimulus could operate in human disease is unknown.
Treatment of MedullaryThyroid Cancer (MTC)
MTC is unique since it can be detected in MEN-IIA or MEN-IIB families by genetic analysis and screening tests measuring pentagastrin-stimulated CT secretion before the disease reaches clinical detectability. In families with familial MTC, MENIIa, and IIB, repeatedly observed elevations to well above the normal range (e.g., peak values of 30-10pg/ml) constitute a basis for operation. In these patients focal tumors or C-cell hyperplasia may be found after near-total or total thyroidectomy is performed. Since C-cell hyperplasia precedes the development of malignancy, it is currently believed logical to operate at this stage of the disease even in young children with MENIIB. Patients with MENIIA or familial MTC are similarly followed, tend to develop tumor later, and 20-40% may never develop cancer (275). International Workshops on MTC have recommended prophylactic thyroidectomy before the age of 5 years in persons harboring mutations of codons 611, 618, 620, and 634 because of their high efficiency in transformation. Consensus on best treatment for patients with other mutations (codons 609,630,768,790,791,804,891) is not clear, with thyroidectomy recommended from ages 5 to 10 (268,276).
Patients with clinical tumor of any size, sporadic or familial, are also treated by near-total or total thyroidectomy. Invasive disease is resected if possible. Since cervical nodal metastasis occur in up to 90% of patients with palpable tumors, a careful mod¬ified radical neck dissection is performed with removal of nodes in the central and ipsilateral compartment. The exploration for nodes should include the upper mediastinum. Bilateral neck dissection may be appropriate in patients with hereditary tumors (277).
Management of Post-operative MTC patients who have positive CT assays
When post-surgical serum CT is undetectable in basal condition and after pentagastrin stimulation, the patient has an almost 100% chance of being in complete and stable remission. In this case follow up should be based on periodic measurement of serum CT, without any instrumental evaluation. After operation, patients with still-detectable serum CT levels should be screened for persistent disease. Imaging should consists of CT of the neck and chest, MRI of the liver and bone scintigraphy. PET scanning may also be informative in selected cases. Some centers used to ablate residual functioning thyroid tissue with 131I, as in papillary or follicular tumors, since this procedure may ablate tiny foci of residual cancer (278,279). However, the value of this procedure is not proven, and this procedure is almost universally abandoned. In patients with local invasive disease, who are above age 45-50, we believe that radiotherapy (5,500-6,000 rads) is useful, although there is disagreement on this point.(v.i.). Occasionally CT levels fall gradually over a year. It is also possible to follow the serum level of CEA as a tumor marker, or CT-related peptide (280). A discordant elevation of CEA in relation to CT may be an indicator of an aggressive tumor. However, in general, CT is the most informative marker.
What to do about the frequently-found elevated CT level without an obvious tumor source is less certain. Residual or recurrent resectable disease should be approached surgically. Neck dissection, if not done previously with a unilateral tumor, should be considered. In the absence of an identifiable source of the CT, careful, extensive, microdissection of the neck and upper mediastinum have been proposed in the hope to eliminate the source of CT (281), but the poor results of such extensive operation have dissuaded many from this approach. Catheterization of the superior vena cava and internal jugulars, with an attempt to localize the tumor by means of multiple venous sampling for CT (242) during pentagastrin stimulation, is possible. The technical success of this approach has been demonstrated, but often the CT level is not reduced to normal, even if operation is subsequently performed on the identified area (282). Radiotherapy may be given to identified non-resectable lesions.
Radiation therapy for MTC
In many cases, no source for the CT is found. Watchful waiting is the approach preferred by some experienced clinicians. Alternatively, mantle irradiation can be recommended. As shown by Simpson (283), medullary tumors are radiosensitive, although a full response may not be seen for many months. This irradiation has been shown to prolong local relapse-free time, but not to clearly improve ultimate prognosis (284). The effectiveness of radiotherapy was evaluated in 24 of 139 patients with medullary thyroid cancer who were given radiotherapy because of advanced local disease at presentation. Only one had normalization of calcitonin, but ten remained free of clinical recurrence. Local relapse was significantly reduced after radiotherapy, but there was no difference in ten year survival between those with and without treatment. These authors believe that radiotherapy does reduce the incidence of loco-regional relapse, but do not advise routine use of radiotherapy because of the relatively favourable long term survival of patients even with elevated calcitonin levels after operation (285). Other investigators question the value of X-ray therapy, contending that patients with MEN-II have multicentric foci from the start, and that radiotherapy may actually worsen prognosis (286). Systemic therapy is reserved for patients with proven symptomatic metastases, and the program is described below.
MTC is associated in MEN-II with parathyroid adenomas and hyperplasia, and in both MEN-IIA and MEN-IIB with pheochromocytomas that are often bilateral and malignant. Parathyroid hormone, VMA, and catecholamine assays should be done to evaluate these problems, and pheochromocytoma, if present, should be treated before the thyroid cancer. Occasionally neuromas cause problems requiring surgery of the intestine or other organs, including the larynx.
BIOLOGICAL FUNCTION OF THYROID TUMORS
Interesting abnormalities in iodide metabolism are typical of thyroid adenomas (described above) and in thyroid carcinomas. In general, the tumors are cold at 131I scintiscan, because they tend to lose the ability to accumulate iodide from the plasma, to bind iodide to TG, or to synthesize thyroid hormone (287). The pattern is one of dedifferentiation, the loss of those functions peculiar to the thyroid gland. Follicular carcinomas tend to retain iodide metabolism more completely than other tumors, and therefore may be susceptible to treatment with RAI. Localization of 131I in tumors, if present at all, is spotty rather than homogeneous.
Occasionally follicular thyroid cancers express high levels of Type2 iodothyronine deiodinase, and cause "consumptive hypothyroidism" (288). This surprising phenomenon has also been observed in patients who have highly vascular tumors which express the deiodinase (289).
In rare cases follicular tumors or their metastases produce significant amounts of thyroid hormone. In many instances the thyroid gland has been completely removed or destroyed, but the metastases have been sufficiently active to maintain the patient in a thyrotoxic state. More frequently, the metastases produce enough hormone to maintain the patient in a euthyroid condition, but not enough to produce thyrotoxicosis. This action indicates that the tumor may be responsive to thyrotrophic hormone, like the observation that thyrotropic hormone can induce growth of the tumors.
Carcinomas usually are associated with elevated serum TG levels. Growing differentiated tumors may cause levels of TG of > 2000 ng/ml. In Hurthle cell tumors and anaplastic cancers, TG levels are usually minimally elevated.
Differentiated thyroid cancers tend to retain a normal affinity and capacity for binding of TSH to their membrane receptors (290), and TSH stimulates cAMP production normally in tumor tissue. Perhaps in keeping with the clinically recognized unresponsiveness to TSH, undifferentiated tumors lack high-affinity TSH receptors. Genetic studies show that in these tumors the expression of the TSH receptor gene, and other thyroid differentiation genes is usually lost (291). In autonomous thyroid nodules, TSH stimulates cAMP accumulation normally; hypersensitivity to the hormone has sometimes been found (292,293).
Immunologic Studies on Tumors
Differentiated thyroid cancers contain TG and microsomal (TPO) antigens cross-reacting with antibody to normal thyroid antigens; this fact can be very useful in identifying metastatic deposits (294). Some tumors tend to lose the microsomal (TPO) antigen (295,296). There is strong evidence that the immune system mounts a defense against the tumors, although an imperfect one. Tumors commonly are infiltrated by lymphocytes, and there is less tendency for nodal metastasis, and a better prognosis, with tumors so involved (207). Antibodies to TG and TPO antigen are more common in cancer patients than in control subjects (254). We have shown, using in vitro assays, that 50% of patients develop immunity to TG, and some react to apparently specific thyroid tumor antigens (297). The tumor antigen is recognized in both autologous and heterologous tumors, and is not present in Graves’ disease tissue. Immunization with tumor preparation has been performed, and in a few cases has led to higher in vivo and in vitro reactivity and apparent partial tumor regression (298). Non-specific immune complexes are found in 10% of patients with differentiated cancers, and specific TG containing complexes are present at low levels in 30% of patients (299). With progress of thyroid cancer, the immune system gradually loses responsivity, as with other tumors (296,298,299).
In vivo, circulating antibodies to Tg and/or TPO are found in nearly one fourth of the patients, and the anti-TG antibodies cause interference in assay of this substance thus making follow-up of patients difficult. Their presence seems not to influence the tumor outcome (300,301). Interestingly, the disappearance of antithyroid autoantibod¬ies during follow-up, correlates with achievement of complete remission, suggesting that elimination of the antigen with successful therapy causes the disappearance of the corresponding autoantigen. Thus, the conversion from antibody positive to antibody negative may be interpreted as a marker of remission, while the persistence of the antigen may be regarded as indirect evidence of persistent disease (302).
The fibrosis and amyloid deposits in medullary cancers might suggest a host antitumor response, and indeed antitumor immunereactivity is detectable in vitro in most of these patients (303).
Sometimes autoimmunity may play an adverse role. Graves’ disease co-occurs with 4-8% of thyroid cancers, whether by chance or through a true association is uncertain. In some patients with the association of thyroid cancer and Graves disease, the thyroid-stimulatory autoantibodies seem clearly to stimulate the growth of the cancer (198,199,200), but overall, it is not clear that the co-occurrence of Graves’ disease causes thyroid tumors to behave more aggressively.
DIAGNOSIS
Most patients with thyroid carcinoma are recognized because of a positive or suspicious FNA done for the clinical or sonographical discovery of a thyroid nodule, either single or in "multinodular" goiter (304). Anaplastic cancers and lymphomas commonly present as a smooth goiter. Stony hardness, fixation to the trachea, and damage to recurrent laryngeal or cervical sympathetic nerves are other important clinical signs. The problems of differentiating carcinoma from other lesions of the thyroid have been reviewed in the preceding discussion of thyroid nodules. A few additional comments are given here. FNA and US studies, constitute the basic and most useful laboratory studies. TSH and fT4 are usually measured to verify metabolic status, and anti-TPO and TG antibodies may be useful in helping differentiate thyroiditis. Isotope scans have a limited role in initial diagnosis.
Thyroglobulin assay-
Although TG assay has been suggested as an important marker for thyroid cancer (305), practice shows that elevated TG levels can be caused by adenoma, multinodular goiter, and other diseases; thus the determination is of little value before operation. Chest xray may be informative but is often omitted. In lesions which extend outside the thyroid, or have metastasis, ultrasound of the neck, CAT scanning of the lungs, and MRI of the neck can provide useful information prior to surgery, and especially when following disease progress.
Fine Needle Aspiration cytology-
Currently, as described previously in the section on diagnosis of thyroid nodules, most reliance is placed on needle aspiration cytology. Fine needle aspiration cytology of cervical nodes under ultrasound guidance also must be remembered as a very useful technique (306).
Ultrasound
Typical features of ultrasonic scanning can suggest malignancy with rather good sensitivity. Microcalcifications, irregular margins, hypoechogeneicity, intranodular blood vessels and round shape are all in support of malignancy and may dictate the need for FNA. The US procedure has replaced other scanning procedures such as angiography, thermography, iodopaque "thyrography", and fluorescent scanning. US is described in the chapter on thyroid nodules and 6c. Very recently thyroid elastosonography has been proposed as a promising new tool to distinguish benign and malignant nodules.
Isotope scanning
Scintiscanning using 131-I is currently usually omitted in the initial evaluation of a possible malignancy. Most of the anatomic information needed is provided by US. Occasionally isotope scanning is useful in demonstration of hyperfunction in a nodule, lack of a lobe, extension below the sternum, or other factors. Demonstrate of failure of the involved area to concentrate RAI used to be important, since malignant lesion are invariably cold at scan. However it in no way proves the presence of cancer, nor does its absence rule out this possibility. Whole body scintiscanning is useful to determine whether lesions in lung or bone are thyroid tumor metastases. Significant accumulation of RAI by the metastasis definitely proves thyroid origin. Occasionally uptake of RAI can be demonstrated in metastases in the neck before surgery. This finding is almost certain evidence for cancer. Usually normal thyroid tissue must be destroyed and TSH elevated before a metastasis will accumulate 131I. 131I whole body scanning in management of previously diagnosed thyroid cancer is discussed later in this chapter. Some tumors accumulate iodide but do not organify it. They are delineated by radiotechnetium scans done during the early "iodide phase" of isotope distribution, but not by scans using131-I or 125-I done at 24 -72 hours, when the storage of organified isotope is primarily recorded. Conversely, some metastases accumulate small amounts of 131-I but are not shown on short-term technetium scans (307).
Recently, PET scan has been introduced and found informative for the imaging of metastatic disease devoid of 131-I uptake. 18-F Fluorodeoxyglucose Positron Emission tomography can localize tumor and determine tumor volume. Large deposits (>125ml) have a very adverse prognostic implication (308). It appears that stimulation of metastatic deposits by elevated TSH makes PET scanning more sensitive (309,310).
CHOICE OF OPERATIVE PROCEDURE
Which operative procedure is indicated when FNA is suspicious or indicative of cancer? (Table 18-6)
Suspicious FNA lesions have a nearly 70-80% chance to be malignant, while an FNA indicative of papillary thyroid cancer is almost always true positive at final histology. Thus, we recommend total (or near-total) thyroidectomy as the initial surgical procedure in these categories, regardless of the size of the nodule. "Near-total" thyroidectomy refers to a procedure which intentionally leaves small portions of thyroid tissue near parathyroid glands or at the entry of the recurrent nerve into the larynx, and is associated with a marked reduction in possibility of hypoparathyroidism and nerve damage. It is frequently used with intended 131I ablation of residual thyroid tissue.
Some authors prefer lobectomy with frozen section examination in case of suspicious FNA. .It must be noted that frozen section carries a high rate of false negative diagnosis, compared to final histology. For this reason, some authors prefer to do total (or near-total) thyroidectomy without performing frozen section. In addition, in many follicular lesions the diagnosis of malignancy can be made only from paraffin sections.
Table 18-6. Suggested Surgical Procedures in Thyroid Cancer
| TYPE | CLASS | OPERATION |
| Papillary, Follicular | 1, <1cm | Lobectomy +/- contralateral STT* (if a < 1cm tumor is detected in a resected specimen, do not reoperate) |
| Papillary, Follicular | 1, >1cm, or multicentric, or post-irradiation | NTT** |
| Papillary, Follicular | II | NTT + MND*** |
| Papillary, Follicular | III | Resection without mutilation |
| Papillary, Follicular | IV | Resection without mutilation |
| Medullary | Any | NTT , MND, see later discussion of extensive node dissection |
| Anaplastic | Any | TT or tumor resection if possible |
* STT = Subtotal thyroidectomy ** NTT = Near-total thyroidectomy *** MND = Modified neck dissection
Among patients with papillary cancer within the gland, some will have cervical lymph node involvement and others will have no obvious spread. The utility of prophylactic neck dissection is controversial. Sone authoritative centers are in favour but other, including the authors of this chapter, prefer to perform central neck dissection only when there is a preoperative evidence of lymph node metastases at US or intraoperative evidence, The same attitude seems indicated for lymph node dissection of other node chains. Whenever a patient treated with lobectomy is found to have cancer at final histology, the question arise whether to perform completion thyroidectomy. The indication of several guidelines (39) are in favour of completion thyroidectomy, with the exception of patients with unifocal, small, intrathyroidal, papillary thyroid cancers without evidence of lymph node metastases and favourable histology.
The approach proposed here, is based on several observations. Multicentric involvement is reported to range from 25 to 90%. The wide variation of multicentricity (or intraglandular dissemination) can be explained in part by the finding that the incidence of multicentricity is doubled if one does whole gland histologic sections. There is little or no relationship between the size of a solitary nodule and the incidence of intraglandular dissemination, but an increasing degree of histologic malignancy is associated with the frequency of dissemination.
Mazzaferri et al., in their review of 576 cases of papillary carcinoma, found that total thyroidectomy significantly reduced the incidence of recurrences, and recurrences will presumably be correlated with deaths from disease (234). Samaan et al (311) also supported this procedure. Hay et al. evaluated the efficacy of different surgical approaches to treatment of patients with low risk papillary carcinoma at the Mayo Clinic and concluded that more extensive surgery was not associated with lower case specific mortality rates, but was associated with a lower risk of local regional recurrence. Their data supports the use of bilateral resection as the preferable initial surgical approach (312).
Total thyroidectomy carries an increased risk of hypoparathyroidism, recurrent nerve damage, and the necessity for tracheostomy (313). Accidental unilateral nerve damage may reach 5%, but fortunately bilateral injury is rare (314). All surgeons attempt to preserve those parathyroid glands that can be observed and spared, and an attempt is often made to transplant resected glands into the sternocleidomastoid muscles. Reports range from a 1 to a 25% incidence of hypoparathyroidism after total thyroidectomy (234,315).
TUMOR STAGING AFTER SURGERY
Tumor staging is intended to identify the risk of death or recurrence after initial treatment. The most used staging system is the TNM Staging system which combines simplicity with rather good predictive power.
Several other staging systems have been developed. One is the Clinical Class system developed at the University of Chicago and is based simply on the extent of disease (316). Other systems are designed to predict outcome. The EORTC classification pro¬posed by the European Thyroid Association is based on age, sex, histology, invasion, and metastases (317). The Dames classification includes data on age, extent and size of primary, distant metastases, and DNA ploidy (318). MACIS includes data on age, invasion, metastases, size, and completeness of surgery (319). All of the systems appear to be effective in categorizing patients into largely similar low and high risk groups.
Several groups have recently established new criteria for risk assessment based on pathological features combined with clinical features and with the response to initial therapy. The idea is to delay the risk assignement to a time when the response to initial therapy may be evaluated. The first proposal (called “Ongoing Risk Stratification”) came from the Memorial Center in New York (320), where patients were assigned to low or high risk category based on the results of follow-up after initial treatment. Patients in apparent complete remission at that time were defined as low risk, regardless of the initial risk stratification obtained soon after surgery. A second proposal came from an Italian study (321). These authors assigned patients to low or high risk group at the moment of the first evaluation done 8-12 months after surgery and radioiodine ablation (if performed). Patients free of disease (negative neck US, undetectable basal and stimulated serum Tg and no other evidence of disease) were classified at low risk. Patients with any evidence of ersistent disease (including detectable TG) were considered at high risk of recurrence. The authors demonstrated that nearly half of the patients could be shifted from the high risk category (at the time of surgery) to the low risk category. The system was named Delayed Risk Stratification (DRS). One advantage of these delayed risk stratification systems is that gives an idea of the risk of recurrence which is not considered in the TNM classification.
TSH SUPPRESSIVE/REPLACEMENT THERAPY
After operation all patients are kept on TSH-suppressive thyroid hormone therapy with l-thyroxine . At the time or the first post-surgical evaluation, individuals with current active cancer (other than medullary or lymphoma) should continue with TSH-suppressive therapy aimed to a TSH around 0.1 µU/ml. Pushing TSH below this level has not been associated with better outcome, while has been associated with more frequent side effects from clinical or subclinical hyperthyroidism. Patients who are considered free of disease, should have their replacement lowered to provide a TSH in the low-normal range, and ultimately as safety is assured, in the normal range.
CHILDHOOD THYROID CANCER
Some special features of thyroid cancer occurring in children deserve comment. It is, of course, an uncommon disease. The tumors are usually papillary or mixed histologically, and tend to grow slowly, with a high frequency (50 -80%) of neck metastases, but with a relatively favorable prognosis. Very young patients (under age 12) often have relatively aggressive disease. The association with x-ray exposure has already been discussed. As in adults, the incidence in girls is double that in boys. Multicen¬tricity of tumors is found in 30 -80%. Metastases to lungs, usually microscopic, are common (perhaps 20%), but tumor is rarely found in the bones. Lung metastases usually accumulate 131-I and can often be eradicated with this isotope, particularly those not visible with X-rays.
As with adult tumors there is no universally accepted surgical approach, but it is certain that sentiment has swung away from prophylactic and radical neck dissections to a more conservative position (322,323). The operations employed are as described above, and near-total thyroidectomy, done by an experienced surgeon, is favored. Thyroid remnants are destroyed with 131-I in patients with multicentric lesions and in all Clinical Stage II, III, and IV. Detection of metastases is attempted by131-I scanning, as described elsewhere in this chapter. Most childhood metastatic thyroid cancers are found to accumulate sufficient 131-I to allow useful and sometimes curative therapy, often with doses of 75-150 mCi. Presumably children are more likely to suffer side-effects of 131-I therapy, as described below.
Reproductive potential is diminished by large doses of 131-I (324), but an increased incidence of birth defects has to date not been encountered among the relatively few progeny studied (325-327). Thyroid hormone is given to suppress TSH to the 0.1uU/ml area in patients who have known residual or probable residual disease, even though this is known to cause some loss of bone mineral. Although the 10-year survival is from 90 to 95%, long term follow-up demonstrates an eight fold greater than normal mortality (230) and emphasizes the need for comprehensive therapy and long term follow-up.
RAI 131-I ABLATION
Most patients who have had a "total" thyroidectomy, and all patients who have had a subtotal resection, will have some functioning thyroid tissue remaining in the normal position after surgery, and will thus be candidates for 131I ablation. This is done to remove any possible residual tumor in the thyroid bed (thyroid ablation), to make subsequent scans and TG assays more interpretable, and (hopefully) to kill tumor cells elsewhere (adjuvant therapy). There is no unanimity regarding the use of postoperative 131I ablation in Stage I tumors, since absolutely convincing evidence of its value is lacking (234,328). But for all patients with papillary and follicular cancers as a group, 131I ablation correlates with improved survival (232). Our data demonstrate that postoperative 131I ablation correlated with decreased recurrences for all patients with papillary cancers over 1 cm in size. Samaan et al (311), in a review of 1599 patients, observed that 131I treat¬ment was the most powerful indicator for disease-free survival.
Ablation can be accomplished in most instances by one dose of 30 mCi 131I, giving the patients about 10 whole body rads (329). In our practice 80% of patients are ablated successfully with one dose of 30mCi, and the remainder require repeat therapy at the time of their second scan. Other clinicians find this dose insufficient, and give 50-150 mCi as an inpatient treatment. In part this difference may depend upon the surgeon, since small remnants of residual thyroid are more easily ablated than large amounts of residual tissue. Low dose (30 mCi) ablation of thyroid tissue after near-total thyroidectomy was recently reviewed by Roos et al. Surveying many studies, they concluded that 30 mCi was as effective as larger doses in inducing ablation, and since it could be administered without hospitalizing the patient, was an appropriate treatment (330). Doses of 100 mCi may provide more certain ablation with one dose (although at the expense of greater patient radiation) but there is little difference between ablation rates with does of 30-75 mCi. There is no data proving that one method or the other provides superior results in terms of survival. We do not routinely use ablation in patients under age 21 with tumors under 1 cm. Patients with tumors above this size, older patients, or those with multicentricity or a history of neck irradiation are advised to take 131I. This practice is followed in most clinics.
The indications for thyroid ablation, based on levels of evidence have been detailed in recent ATA guidelines (39). Three groups of patients are identified, one (at very low risk of recurrence) in which thyroid ablation is not indicated due to the lack of evidence of any benefit; a second group where the benefit, if any, are not evidence based. In this group, ablation should be offered in selected cases according to the judgement of the treating physician. Finally, a third group, including high risk patients, in which ablation has a strong indication based on good evidence that it may reduce cancer recurrence and possibly deaths.
Irrespective of the protocol and the dose used for ablation, there is always a subgroup of about 20% of patients that will not be successfully ablated with the first RAI course. The factors associated with ablation failure are not fully understood. Ablation failure does not correlate precisely with the dose, with the levels of TSH stimulation, the amount of thyroid residue or the level of urinary iodine excretion (331). In particular, it is not certain whether the use of doses higher than 3.70 GBq would result in any addi¬tional benefit, or whether there is a ’stunning’ effect of the diagnostic dose of 131I on the subsequent ablation rate, although likely to occur. A retrospective analysis was performed of all patients (n=389) with well-differentiated thyroid cancer treated at our institution between 1992 and 2001. The therapeutic dose was the only variable found to be associated with success (odds ratio, 1.96 per 1.85 GBq increment). Our results confirm the presence of a significant percentage of ablation failures (24.4%) despite the use of high ablative doses (3.70-7.40 GBq). Higher therapeutic doses are associated with higher rates of successful ablation, even when administered to patients with more advanced stages. Higher diagnostic doses were not associated with higher rates of ablation failure. (332).
The utility of radioactive iodide treatment of patients with papillary and follicular cancer was recently reviewed in a series of articles by Wartofsky, Sherman, and Schlumberger and their associates. Schlumberger concludes that routine radioactive iodide ablation is not indicated in patients with differentiated thyroid carcinomas of less than 1.5 cm in diameter, and advocates restricting RAI ablation to patients with poor prognostic indicators for relapse or death (333). Wartofsky points out a secondary benefit of postoperative low dose 131I ablation in that, for many patients, it provides a high degree of certainty and peace of mind when subsequent scans are negative and TG is undetectable. Another argument for radioactive iodide ablation and early detection of any recurrence is the data presented by several groups, including Schlumberger and colleagues, that there is a reciprocal relationship between the success of cancer therapy and the size and duration of the lesions.
In patients with Stage II to IV disease, we proceed to destroy all residual thyroid and to treat demonstrable metastases if they can be induced to take up enough 131I. Use of 131I therapy is investigated in these patients, regardless of the histologic characteristics of the resected lesion, although significant uptake rarely is found in Hurthle tumors (243,334) or in patients with anaplastic lesions.
Preparation for 131-I ablation
The "traditional" approach has been to induce hypothyroidism prior to the ablative dose in order to raise TSH and stimulate uptake of RAI in residual thyroid or tumor. This may be done by simply leaving the patient without T4 therapy for 3 weeks post op. Alternatively patients can be given thyroid hormone suppressive therapy for 6 weeks or so after operation, so that any malignant cells disseminated at the time of thyroidectomy will not be stimulated by TSH. The value of this measure is admittedly unknown. Patients then receive 25 µgL-T3 bid for 3 weeks, and therapy is then stopped for at 2-3 weeks to allow endogenous TSH (which may reach 20-60 µU/ml) to stimulate uptake of the 131I by the remaining fragments of thyroid tissue or metastatic lesions in the neck or elsewhere before proceeding with 131I therapy. Diagnostic scans are no longer indicated by several groups and ATA guidelines (39), based on the evidence that they do not offer additional information compared to the post-therapy scan and based on the possibility of stunning. However, if one wants to do it, the usual scanning dose should be no higher than 1 mCi 131-I, and scans are read at 48 or 72 hours, when body background has diminished. If TSH is sufficiently elevated the initial scan can reveal distant metastases as well as residual thyroid gland. If large thyroid tissue remnants are present, TSH may not become very elevated, but will do so after the first ablation dose. Patients with Class I and Class II disease under age 45 are given 30 mCi as an out-patient treatment. Older patients with Class II disease and patients with Class III or IV disease are given doses of 75-100 mCi as an inpatient treatment. A post-therapy whole body scans should be mandatory 3-5 days after the ablative dose of 131I (or after therapeutic doses), since occasionally unsuspected metastasis may be visualized on scans at this time. Serum Tg is always measured at the time of 131-I therapy. At 24 hours after initial ablation, we replace hormone therapy at suppressive doses
Some physicians proceed without prior scanning directly to 131-I ablation 2-4 weeks after surgery and perform a post-therapy scan 5-7 days later. Presumed benefits of this approach are patient convenience, less expense, and avoidance of possible thyroid "stunning" by the scan dose. In fact, stunning has not been demonstrated with the 2mCi 131-I dose, although it may occur. Arguments for doing a pre-ablation scan include finding out the actual percent uptake of the treatment dose in the neck and elsewhere, establishing if in fact there is uptake, and recognizing disease that may dictate a larger initial dose. The final word on these different approaches is not in. Variations on this approach were studied by Pacini et al (335), who compared induced hypothyroidism with rhTSH stimulation. The Pisa group found that either thyroid hormone withdrawal, or hormone withdrawal plus 2 doses of rhTSH, produced higher percentage uptakes and more frequent ablation with 30 mCi doses (in about 80% of cases), compared to rhTSH alone. These results have been confirmed in other series (336), including a randomized, international study (337) which brought the approval of Thyrogen in the preparation of thyroid ablation.
Half-Dose Protocol and Thyroid Hormone Withdrawal
An alternative to rhTSH stimulation for the initial follow-up is the "half-dose" protocol (338). Half the usual dose of thyroxine is given for six weeks. TSH is tested in the fifth week, and if over 20 uU/ml, scanning is done in the sixth week, or preparation is prolonged if needed. On this protocol patients usually feel quasi-normal and conduct normal activities, in contrast to their function during withdrawal. On the half-dose protocol, FT4 falls to just below normal, and TSH on average reaches about 60uU/ml in the sixth week. Patients who start with TSH below 0.1 U/ml may take longer to reach a satisfactory level for Tg testing, which is generally considered to be with TSH at least 30 U/ml.
Recombinate human TSH (Thyrogen)
During induced hypothyroidism, patients may experience a wide range of hypothyroid signs and symptoms which may be severe and may result in a substantial impairment of the patients’ lives and ability to work, and occasional tumor growth. Recombinant human TSH (Thyrogen) has been developed to meet the need for safe, adequate exogenous TSH stimulation in patients with papillary and follicular thyroid carcinoma. In vitro studies have shown that rhTSH can effectively stimulate cAMP production and the growth of human fetal thyroid cells. The in vivo biological efficacy of rhTSH was demonstrated in normal subjects, in whom it is able to increase serum T4 and T3 concentrations and stimulate thyroidal radioiodine uptake. A single dose of 0.1 mg rhTSH is a potent stimulator of thyroid function in normal subjects (339).
A first clinical trial of recombinant human TSH (rhTSH-THYROGEN) (phase I/II) was completed in 1994 in 19 patients after a recent thyroidectomy for differentiated thyroid cancer (340).The encouraging results of this limited study were confirmed in a larger multicenter phase III study conducted between 1992 and 1995 in the USA in 127 patients (341) and in a second phase III multicentric trial, which included USA and European centers (342). The results of this trial can be summarized as follows: scans were similar after rhTSH and thyroid hormone withdrawal in 92% of the patients, with no difference between the two dose regimens investigated. When the results of 131I WBS and post-rhTSH Tg levels were combined, the detection rate increased to 94%. Among the patients with persistent or recurrent disease, 80% had concordant scans, 4% had superior rhTSH scans and 16% had superior withdrawal scans. Interestingly, serum TG levels were detectable in 80% during thyroid hormone therapy and were detectable in all following either rhTSH stimulation or withdrawal of thyroid hormone treatment. However, the TG level reached after rhTSH stimulation was in general lower than that obtained after thyroid hormone withdrawal. Tissue RAI uptakes obtained in the patients undergoing hormone withdrawal were twice the values found after rhTSH, indicating that withdrawal provided a much greater stimulus to thyroid or tumor tissue. However, as noted, the diagnostic results were nearly equal. Quality of life was much better during rhTSH than during hypothyroidism induced by thyroid hormone withdrawal, and side effects were minimal, mainly consisting in mild and transient nausea or headache in less than 10% of patients. No patient has developed detectable anti-rhTSH antibodies, even after receiving repeated courses of rhTSH in successive clinical trials .All together these clinical trials have clearly shown that rhTSH is an effective and safe alternative to thyroid hormone withdrawal during the post-surgical follow-up of papillary and follicular thyroid cancer, although not as sensitive as scanning after hormone withdrawal in some patients. Another factor to consider is the cost, wehich is rougly $ 2000 per treatment, although for the majority of patients in USA this is covered by their insurance. A few patients have been reported with metastases demonstrated on withdrawal scans that were not evident on rhTSH scans (343). It has been found that Thyrogen administration induces a reduction of serum vascular endothelial growth factor, even in the absence of thyroid tissue (344). The clinical significance of this observation, if any, is unknown, but it does imply possible action of rhTSH on receptors other than in thryoid tissue. Use of rhTSH in managing thyroid cancer has recently been extensively reviewed (345). Thanks to many studies confirming the properties of rhtsh in stimulating iodine uptake and Tg production, rhTSH has now considered a standard method of preparation for both thyroid ablation and post-surgical follow-up in patients with any form of differentiated thyroid cancer.
Options in Follow-up scans and treatment-including recently described variations
After surgery and thyroid ablation, the next step is follow-up. The first important time for follow-up is between 8 and 12 months after initial treatment. At this time we want to understand whether the patients have evidence of complete remission or some evidence of persistent or recurrent disease. In the past, the conventional preparation for follow-up was to obtain a diagnostic total body scan with 131I after induction of hypothyroidism, with the same methodology as described for ablation, in order to stimulate uptake of 131-I by residual thyroid tissue or tumor cells and production of TG. In recent years it has become common to omit the diagnostic scans after initial ablation, at least in patients deemed to be at low risk, and relying entirely on measurement of stimulated (after rhTSH administration) serum TG when anti-Tg antibodies are negative (39). In patients known to have residual disease because of elevated baseline TG or ultrasound evidence of metastatic lymph nodes, is to give therapeutic 131-I without preliminary scanning. In several large series, it was demonstrated that at this time of the follow-up, more than 80% of the patients will have evidence of complete remission (negative neck US and undetectable stimulated serum Tg levels). These patients do not require additional tests or imaging and their suppressive hormone therapy should be shifted to replacement targeting serum TSH in the low-normal range. In subsequent years, the chance of these patients to have a recurrence is extremely low (<1%) and thus their follow uo should be solely based on basal Tg measurement and neck US once a year. On the contrary, when neck US is positive for local disease, or the basal or stimulated Tg is elevated the patient should be screened for the localization of the disease and treated accordingly.
FOLLOW -UP TREATMENT BASED ON TG ASSAYS
As assays for thyroglobulin (TG) have become more sensitive and reliable, measurement of TG assumes more and more importance in determining the management of patients followed after thyroidectomy and radioactive iodide ablation treatment for thyroid cancer. Serum TG levels, in the absence of antibodies interfering in the assay, correlate well with tumor burden, although detectable tumor may well be present even in the presence of negative TG assays in individuals who are on replacement thyroid hormone (346). Pacini et al (347) reported that in a retrospective study of 315 patients who had undetectable serum TG in the hypothyroid state at the time of the first control body scan after thyroid ablation, no useful information was provided by the body scan. Thus they propose that diagnostic 131I whole body scans can be avoided in patients with undetectable levels of stimulated TG after initial ablation, and that the patients can be monitored with clinical examination, ultrasound, and serial TG measurements on thyroxin treatment during the subsequent follow-up.
However, some concerns may be noted. The TG assay used in their study recognized a value of <3ng/ml as "undetectable". Also, of the 90 patients in the study with thyroid bed RAIU, 54 received a second course of treatment, and seven received two additional courses. This seems to question the recommended approach (347). In a second study (348) they found that an undetectable TG, when hypothyroid at the time of the first control scan after ablation, predicted complete and permanent remission. They propose that subsequent 131-I scanning is unneeded, and follow-up clinically and by TG assays while on thyroxin, would be sufficient. Mazzaferri and Kloos (349) studied retrospectively 107 patients who were “clinically free of disease” and had undetectable or very low serum TG levels during thyroid hormone therapy. The Tg levels on treatment were all 1 ng/ml or less, and 95% were under 0.5 ng/ml in their assay, which was a commercial (Nichols Institute) chemoluminescent antibody assay. In response to the administration of two doses of recombinant TSH and assay of TG on samples taken on the fifth day, 20% were found to have a value above 2, with values ranging from above 2 up to 18 ng/ml. Twenty percent of the patients who had low or undetectable Tg had elevation above 2 ng/ml after rhTSH stimulation, and many of these patients ended up with therapy. However, the authors found that radioactive iodide whole body scans often failed to localize the source of the elevated TG, which was found after post-therapy scans or by other imaging methods. This study suggests that even with a TG level below 1 while on replacement therapy, persistent disease may sometimes be present and be detected by stimulation using recombinant TSH or thyroid hormone withdrawal.
Wartofsky (350,351) comments on these studies and supports the idea that TG testing, both on suppression and after TSH stimulation, can help in determining therapy. He suggests that, in patients with a serum TG<0.5ng/ml on suppression, and in a low risk category, that stimulation by recombinant TSH and measurement of TG, rather than scanning, is satisfactory. If the TG remains <1, the patients can be evaluated annually with such a stimulation test. In patients with slightly higher TGs, up to 2, he suggests measuring a recombinant TSH stimulated TG, and scanning. In patients with higher TGs, he suggests that thyroid hormone withdrawal and radioactive iodide treatment, without initial scanning, may be appropriate. In a study done by the rhTSH-Stimulated Thyroglobulin Study Group (352) and published in 2002, a cut off level of 1 ng/ml for stimulated TG was taken as the safe level for patients with low risk. This group would presumably be monitored by repeat rTSH stimlated TG assays rather than scans. It is of interest that in this study 14 of the patients with stimulated TG <2ng/ml underwent isotope scanning and 9 were positive. These "could be interpreted as false negative" tests. Five had uptake outside the thyroid bed. This group suggests that patients with stimulated TG above 2 would have subsequent thyroid hormone withdrawal and possible 131-I therapy without scanning. A recent “consensus” statement by a group of thyroidologists also supports the categorization of patients into high and low risk groups, and use of TG as described above for following low risk patients (353).
Whether this approach, with omission of whole body scans, has any adverse effect on long term outcome is not yet known.
STUNNING FROM "TRACER" RAI-
The amount of 131I used for scanning varies from 2-10 mCi, in various clinics, or even larger amounts. Clearly the larger doses detect more lesions, but this rarely alters treatment plans. More importantly, doses of 5-10 mCi have been shown to decrease tumor uptake of the subsequent treatment dose due to "stuning" of the tumor (354). The exact importance of this phenomena is uncertain, but use of a 2mCi dose has seemed to be a reasonable compromise. A study by Lassmann et al (355) call this assumption into question, since they found that even a 2mCi scan dose reduced RAIU by up to 50% in a second procedure done 6 weeks later.This study was carefully performed on a few patients, but it is possible that prolonged stimulation of the thyroid by elevated TSH may have adversely altered iodide kinetics in the remnants during the second RAIU procedure.
Although a reduction of uptake after prior low dose scans has been reported, it is uncertain that this limits the effectiveness of treatment. It was found that ablation rates were equal in individuals who had, or had not, received scans prior to their treatment dose of 131-I (356).
Ingestion of large amounts of iodide, or exposure to contrast dye within 6 (or more) weeks, can prevent RAIU. Uptake can be enhanced by prescribing a low iodine diet for at least two weeks before scanning and therapy, although also this assumption has been questioned by a recent study (331). It is useful to give magnesium citrate to induce bowel emptying prior to the scan. As discussed above, the value of routine follow-up diagnostic scans has been challenged by two European study, who suggest that such scans rarely give information of value in patients who have TG <1 after thyroid ablation (357,358). They suggest testing TG after thyroid hormone withdrawal or rhTSH, and directly treating those with TG above 10 by administration of 100mCi 131-I. This concept is of interest and awaits further examination, but has obvious problems as noted.
HIGH DOSE RAI THERAPY FOR INVASIVE OR METASTATIC DISEASE
Efficacy and morbidity of high activity of 131-I therapy was assessed in 38 patients with locally advanced or metastatic differentiated thyroid cancer (16 follicular, 20 papillary, one Hurthle cell, one insular) who were treated with high activity radioiodine therapy (9 GBq) as the cancers had previously not responded to standard activities of 5.5 GBq.
After high activity treatment, 9.7% of patients suffered grade 3 and 3.2% suffered grade 4 WHO haematological toxicity. Significant salivary gland morbidity was observed (30% dry mouth, 27% salivary swelling). In this study repeated treatment with high activity (9 GBq) in patients with advanced differentiated thyroid carcinoma appeared to be of no apparent benefit and lead to late morbidity (359). However, other investigators have differing results. A retrospective analysis was conducted on 124 differentiated thyroid cancer patients who underwent dosimetric evaluation using MIRD methodology over a period of 15y. One hundred four RAI treatments were performed. A complete response at metastatic deposits was attained with absorbed doses of >100Gy. No permanent BM suppression was observed in patients who received absorbed doses of<3Gy to BM. The maximum administered dose was 38.5 GBq (1,040 mCi) with the BM dose limitation. Dosimetry-guided RAI treatment allowed administration of the maximum possible RAI dose to achieve the maximum therapeutic benefit. Estimation of tumor dose rates helped to determine the curative versus the palliative intent of the therapy (360).
131-I Treatment using empirically determined doses of 100-250mCi
Patients who have significant uptake of 131I in metastases (usually above 0.5% of the tracer) are given 150-250 mCi 131I. This dose can be tolerated without acute radiation sickness, and is below the level that would promote pulmonary fibrosis if diffuse pulmonary metastases are present, unless uptake in the lungs exceeds 50% (see below). Although use of these empirically derived doses is the most common practice, some centers do careful dosimetry with a tracer dose of 131-I prior to therapy, in order to judge the appropriate, or maximal safe, dose. This requires 2-5 days of observation. The methodology and results have been recently discussed (361). Whether administration of maximally large individual doses is more effective than use of somewhat smaller doses of 131-I has not been established. In perhaps four-fifths of patients accumulating 131I, it is possible to administer a dose of RAI that should be useful in destroying tumor. For normal thyroid tissue 10,000-15,000 rads is destructive, and a dose of 20,000 rads or more is probably needed for therapy of cancer. Assuming, for example, a standard 150 mCi 131I dose, and delivery to tumor of about 100 rads per micro Curie retained per gram, a 1% tumor uptake distributed through 10 g of metastatic tissue could provide an effective treatment.
Some groups have attempted to measure tumor volume by use of quantitative PET scanning (362). The effective half-life can be determined from serial counts of the tracer over the metastasis. If 10 g of tumor in the neck accumulated 1% of a 150 mCi dose, and isotope turnover in the tumor was extremely slow, the radiation dose might be as follows: Rads =74X0.19X 150,000X0.01X6 / 10= 12,654 rads.
The question of whether a sub-cancericidal dose should be delivered in patients with low levels of tumor isotope accumulation needs further investigation, since radiobiologic studies suggest that radiation could preferentially spare the more radioresistant cells, ultimately leaving a more lethal tumor. It may be possible to give conventional x-ray therapy after 131I in those instances in which 131I uptake is present but the total dose delivered to the metastasis is less than adequate. This procedure may provide another therapeutic approach to the thyroid cancer patient, but it has not yet been given adequate trial. Maxon et al (363) report that radiation doses of at least 30,000 rads for thyroid ablation, and 8,000 for therapy to metastasis, improve the rate of response.
Lithium was found to be a potent adjuvant in 131I therapy of metastatic well differentiated thyroid cancer. Koong et al administered 600 mg of lithium carbonate orally followed by 300 mg three times daily and adjusted the dose to maintain a lithium concentration of 0.6 – 1.2 mEq/L, which is effective in blocking 131I release from the thyroid. The estimated 131I radiation dose to metastatic tumor tissue was increased on average 2.29-fold. It is possible that lithium would be of value in managing a large fraction of patients treated with radioactive iodide. It must be noted that lithium has a narrow therapeutic index and that serum concentrations must be monitored. Care must be taken in patients with reduced renal function. It would be imperative in patients who are to receive "maximal 131I therapy" that the measurement of 131I retention in blood and body be performed while the patient is receiving lithium prior to therapy. The use of lithium might also reduce the required 131I dosage for ablation of thyroid remnants (364).
It is useful to do a scintiscan on patients who have received therapeutic doses of 131I at 5 -7days following the treatment, thus using the treatment dose as a more pow¬erful scanning dose. While often offering no new information, this may reveal unsuspected metastasis, especially in younger patients who have previously had 131I treatment. Fatourechi et al found that 13% of follow-up scans demonstrated abnormal foci of uptake not seen on diagnostic scans, and changed management in 9% of their patients (334,365).
The 131-I treatment cycle is repeated at 24-52 weeks, as long as there is no evidence of systemic radiation damage, and as long as the metastases continue to accumulate iodide. The total 131I dosage may vary from 150 to (rarely) 2,000 mCi. Both papillary and follicular cancers respond to 131-I therapy. Small metastases from papillary cancer, especially if functional in the lungs but not large enough to be visualized on X-ray, are typically cured. Follicular tumors often have relatively few metastases and high uptake, thus seem ideal targets for therapy. However portions of the metastases, especially in bone, appear to be resistant and finally continue growth despite 131-I treatment. Nevertheless 131-I therapy is beneficial even in advanced and aggressive tumors. Pelikan et al report their experience on the use of radioactive iodide in treating advanced differentiated thyroid carcinoma and report that up to 50% of patients who have distant metastases can be cured by 131I therapy (366). Aggressive high dose radioiodine therapy has been advocated for treatment of advanced differentiated thyroid cancer by Men-zel and colleagues. These physicians gave repeated doses of 300 mCi (11.1 GBq 131I) with mean accumulated total activities of, on average, 55 GBq per patient. Repetitive high dose therapy appeared beneficial in the majority of patients with papillary carcinoma, but the majority of follicular thyroid cancer patients had progressive disease despite treatment (367). The National Thyroid Cancer Treatment Cooperative Study Registry Group recently evaluated the therapy of high risk papillary and non-Hurthle cell follicular thyroid carcinoma. The study confirmed the utility and benefit of radioactive iodide therapy to reduce recurrence and cancer-specific mortality among patients in the high risk group (368). Pittas et al. (369) reviewed an extensive series of 146 patients with documented bone metastasis from thyroid carcinoma seen at Memorial Sloan Kettering in New York City. Bone metastases were most common in vertebrae, pelvis, ribs, and femur, and multiple lesions were present in more than half the cases. Overall ten year survival rate was 35%, and from diagnosis of initial bone metastasis, was 13%. Favorable prognostic signs for survival included radioiodine uptake by the metastases and absence of non osseous metastases. Hurthle cells had a favorable response to treatment, rather surprisingly, whereas un-differentiated thyroid tumors fared the worst.
Arterial embolization has been combined with radioactive iodide treatment for management of large bone metastasis from differentiated thyroid carcinoma with apparent improvement in effect over the use of radioactive iodide alone. In the study by VanTol et al, (370) embolization was not accompanied by any severe complications.
rhTSH is now available for routine use, and allows 131I therapy without induction of hypothyroidism (340). This will probably increase acceptance of scanning and therefor increase the frequency of the procedures. Iodide depletion by dietary control and diuresis, including furosemide or mannitol administration, can also double the fractional uptake of 131I in metastases (371,372). Finally, when the diagnostic scan shows no 131I uptake, even with TSH, the potential benefits from this mode of therapy have been probably exhausted. However, before giving up to 131-I therapy, some authors suggest using high doses of 131-I and obtaining a post-therapy scan, which in some cases may show areas of uptake not seen in the diagnostic scan (see below).
131-I Therapy with "Negative" scans
In some patients tracer studies fail to show uptake, and serum TG is elevated. Some investigators recommend treating these individuals with large doses of 131I (100¬-150 mCi) and report that tumor uptake can be visualized after treatment, and that serum TG may fall (373,374) (Fig. 18-18). The clinical efficacy of this approach is not known. In a few cases reported by Schlumberger et al. (375) and Pineda et al. (376). TG became undetectable, which clearly is a striking and hopeful result. As of this date, there is no data proving that this treatment improves prognosis (377).
The utility of radioactive iodide treatment of patients with papillary and follicular cancer was recently reviewed in a series of articles by Wartofsky, Sherman, and Schlumberger and their associates (333). Sherman and Gopal analyzed the use of 100 mCi doses of 131I for treatment of scan negative TG-positive patients and conclude that this must, at this point, be considered an experimental procedure of uncertain benefit. They argue against its use in young patients with elevated although apparently stable TG values and without radiographic evidence of disease. Fatourechi et al. (378) analyzed results of this treatment in a series of patients treated at the Mayo Clinic and concluded that it rarely produced significant effect, although it possibly helped stabilze disease in patients with micro metastases in the lung. It is clearly ineffective in patients who have metastases large enough to be detected on chest X-ray of CAT . Wartofsky et al. (333) sug¬gests that, rather than initial treatment with 131I of patients who are scan negative and TG-positive, thorough imaging studies are appropriate. These might include a CAT scan of the chest, an MRI of the neck, 99mTc-MIBI, or 18-fluorine fluorodeoxyglucose PET scanning, or even 99mTc-tetrafosmin, or 201TI thallium. Localization of malignant tissue by any of these means may allow surgical excision or external radiotherapy. This series of articles provides many very useful thoughts on management of diffi¬cult patients with recurrent thyroid carcinoma.
Figure 18-18. Changes in serum TG after therapeutic doses of radioiodine in patients with negative basal whole-body scans. The arrows indicate the administration of therapeutic doses of 131-I. The numbers represent individual patients. In most patients, thyroglobulin decreased, although only in a few to a level that would indicate absence of residual disease. (From Pacini et al, J Nuc Med 28:18888-1891, 1987)
Maximal dose protocols
The therapeutic protocol used at Memorial Hospital in New York, by Maxon (363), and as well at some other centers, has for years been designed to give maximal-tolerable radiation doses to cancer patients (231). The dose is calculated on the basis of prior isotope tracer kinetics. The aim is to give a blood dose of under 200 rads, or less than 120 mCi retained at 48 hours, or 80 mCi retained at 48 hours if diffuse lung metastases are present. This method has theoretical advantages since it potentially provides the most cancericidal dose, but the difficulties of calculating the dose and the occasional adverse reactions have so far prevented this method from being generally employed. The dosimetric approach has been carefully reviewed by Van Nostrand et al (361).
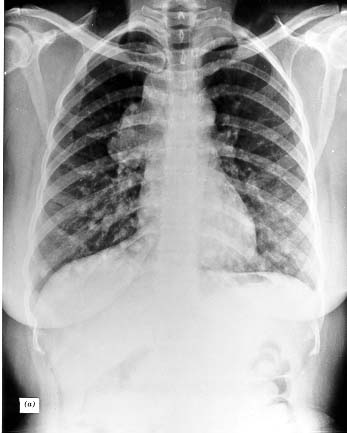
Figure 18-19a. Chest radiographs of a patient with extensive follicular thyroid carcinoma prior to (upper) and after 131-I therapy (lower). With 131-I therapy there was resolution of the large pulmonary metastases, but a tumor in the cervical vertebrae progressed and caused instability of the spine.

Figure 18-19b. Chest radiographs of a patient with extensive follicular thyroid carcinoma prior to (upper) and after 131-I therapy (lower). With 131-I therapy there was resolution of the large pulmonary metastases, but a tumor in the cervical vertebrae progressed and caused instability of the spine.
RADIATION PRECAUTIONS
Before radiation therapy, female patients should be carefully screened for pregnancy and lactation. Confirmed or possible pregnancy constitutes a firm contraindication to therapy because of the risk of damage to the fetus.
A patient who has ingested many milliCuries of 131I can cause serious radiation contamination, and appropriate precautions must be followed. If less than 30 mCi 131I is given, it is permissible to have the patient dispose of urine and feces into general sewage. If amounts of 131I greater than 30 mCi are given, the patient should be kept in a private room in the hospital until less than 30 mCi is retained in the body. Urine can be directly disposed in sewage, or can be collected by the patient and stored in bottles behind protective lead shielding. After physical decay, usually after about 6 weeks, it may be discarded in the sewage. Contaminated bedding and utensils should be stored for 10 half-lives (80 days), thoroughly washed, and monitored for residual contamination before being used again. Alternatively, disposable bedding and utensils may be used.
Table 18-9. Radiation Exposure to Personnel During Care of a Patient Who Has Received 100 mCi 131I
| Distance From Source – e.g The Patient | Reason for Exposure | Rate (mrad/hr) | Allowable Duration of Exposure Permitted on Basis of 0.1Rad/Week |
| 1/2 in. | Direct handling of therapy dose or urine after therapy | 136000 | None |
| 1 ft. | Giving personal hygiene to treated patient | 240 | 0.5 hr/week |
| 3 ft. | Making the bed, mopping the floor | 27 | 5.0 hr/week |
| 9 ft. | In chair across the room | 3 | 50.0 hr/week allowable exposure cannot be reached |
Personnel caring for a patient who has received 131I therapy are often concerned about exposure to excessive radiation. This is almost never a real problem. Isotope can, at a practical level, only be passed from the patient to another person via saliva or urine. Monitoring by means of a portable counter is important in making certain that no person receives more than an allowable radiation dose from the isotope in the patient’s body. Table 18-9 gives a rough estimate of the amount of radiation received while performing ordinary hospital tasks at various distances from a patient who has received 131I. In general, all ordinary patient care can be performed without hazard. It is best to avoid close contact between hospital personnel and patient during the first 48 hours after therapy because of undue apprehension that may be induced. However, even after doses of up to 100 mCi, normal personal actvities such as eating at the same table, or driving in the same car, carry no risk to others.
The US Nuclear Regulatory Commission has published new guidelines which allow release of patients treated with isotopes from the hospital if the total effective radiation exposure from the treated person to any other individual is not likely to exceed 5mSv (0.5 rads). Grigsby et al (379) found that when using precautions such as those described above in a group of patients given on average about 100mCi 131-I, the exposure to other individuals in their household and to pets did not exceed this level. Recently, guidelines for the optimal radiation protection after treatment with 131-I have been proposed by the American Thyroid Association (380).
Radiation damage from 131-I Therapy
The use of RAI in large doses is not without hazard. The radiation dose delivered to the whole body, the gonads, or bone marrow is usually assumed to be the same as that of the blood. The blood dose depends on the amount of isotope administered; its distribution space and turnover; the degree of heterogeneity of distribution in the tumor; the uptake, synthesis, and secretion of labeled compound by the tumor; and perhaps other variables. The radiation is usually largely due to inorganic iodide, since little protein bound 131I ordinarily appears in the blood. Sometimes tumor destruction is such that much PB131I appears in the blood and can yield a major fraction of the total whole body radiation dose. As a rough estimate, the blood, gonadal, or bone marrow radiation may be assumed to be 0.3 -1.5 rads/mCi 131I administered (381), or 45-150 rads per treatment with 100 mCi. The genetic risks are discussed in Chapter 11 and are not reviewed here. Ordinarily, when 131I therapy is needed for carcinoma, the necessity of treating the patient outweighs the risks of genetic damage.
Various unwanted effects of radiation may occur in patients receiving large doses of 131I. Mild radiation sickness is seen. Metastatic deposits or surrounding tissues may become painful over 2-4 weeks from radiation-induced inflammation. Damage to the salivary glands can cause sialadenitis, and xerostomia, and can lead to loss of teeth (382). Increasing salivary flow following treatment is partially protective. Ovarian function is often temporarily suppressed (383), and if there are pelvic metastases that collect 131I, the gonads may receive a sterilizing dose. Sperm count may be reduced for months (384). Leukemia occurs with increased frequency in patients who have received large doses of 131I (usually>600 mCi) for cancer (217). Transient or permanent alterations in liver function and lymphoma of the parotid gland have been reported as possible sequelae (385). Pulmonary fibrosis has occurred in patients with functioning lung metastases who have received unusually large doses or who have very active metastases (386). Leukopenia, thrombocytopenia, and anemia are encountered with accumulating doses. A mild effect on the bone marrow is seen with each therapeutic dose, and after several hundred milliCuries, aplastic anemia may develop (387). The hemoglobin level, white cell count, differential count, and platelets should be monitored periodically in order to judge recovery of the marrow between treatments and to prevent excess total radiation damage to the marrow. Large radiation doses may cause transient swelling of metastasis in the brain or spinal canal.
Lin et al (388) recently reviewed pregnancies following 131I treatment of well differentiated thyroid carcinoma among a group of 58 pregnant women and found no evidence of demonstrable adverse effects, but suggest that it would be wise to avoid pregnancy during the first six months after the last administration of 131I. With the exception of possibly increased rate of miscarriages, no other adverse effect of radioiodine has been found on the outcome of 2113 pregnancies after radioiodine treatment and on their offspring (318).
Two special complications need be noted. Occasionally withdrawal of hormone suppression, in preparation for isotope therapy, leads to rapid growth of the tumor, and reinstitution may not seem to return the patient to the prior condition. Special care should be taken if metastases are present in areas such as brain or spinal column, where growth could cause serious sequelae. Glucocorticoids are occasionally given prophylactically in an effort to prevent tumor swelling in this situation .
RAI was introduced into the treatment of thyroid carcinoma with the hope that it would be a panacea for this disease. Unfortunately, the results have not been universally beneficial. Most tumors in children appear to be treatable, and among adults 80-90% of metastatic carcinomas accumulate sufficient 131I to warrant a serious therapeutic trial. Patients who harbor this form of the disease are fortunate, since 131I may totally eradicate the metastases. Even multiple pulmonary metastases occasionally disappear after 131Itherapy (Fig. 18-19a, above). The final value of 131I therapy has been difficult to define, largely because of a lack of controlled series and because of other treatment (especially thyroid hormone) given at the same time (387). Mazzaferri (232) found that 131I ablation and therapy significantly improved the prognosis in papillary cancer by decreasing recurrences. Varma et al. (389) found that RAI treatment had no effect on the survival of persons under age 40 but did lower the death rate of patients over age 40. Leeper (390) concluded that 131I treatment appears clearly to benefit patients under 40 years of age with papillary cancer, but the course of this cancer in older patients is rarely affected; follicular cancers in older patients are treatable, and survival is prolonged even if the disease is not eradicated. Soft tissue lesions, especially of the lung and mediastinum, respond best to 131I. Osseous lesions are often highly functional but are infrequently totally destroyed by 131I (Fig. 18-9b) (391). Lesions detected on whole body scans, with negative bone X-rays, are most likely to be cured. In another report, 59 of 400 patients were considered candidates for 131I therapy after using antithyroid drugs or TSH to stimulate uptake. Of these, 61% with metastatic disease were benefited (362). The follicular, papillary, and mixed cancers responded equally well. Numerous reports indicate that ablation of metastasis with 131I is associated with a better prognosis than failure to ablate, but obviously this outcome may relate to the histologic nature and function of the lesion rather than to the therapy per se.
Disseminated pulmonary metastasis can sometimes be eradicated by 131I, but radiation pneumonitis or fibrosis may be produced and may be fatal (321,386). On first observation of pulmonary metastases, this therapy should be considered, but no more than 75 mCi should ever be deposited in the lungs in one treatment. Progress of the lesion and pulmonary function should be carefully evaluated before and be-tween treatments (389,391,392-394). Occasionally patients present with locally advanced papillary thyroid cancer which is not surgically resectable. In some instances preoperative treatment with radioactive iodide sufficiently reduces the extent of the lesion to allow subsequent definitive surgery (395). One of the most informative study regarding the effects and limitation of 131-I therapy is given by the series of the Institut Goustave-Roussy periodically updated by Schlumberger et al (396). In their most recent publication the authors report on 444 patients treated with 131-I for distant metatsases and the results identified three groups of patients with different outcome after therapy: a group very likely to be cured after a few courses of RAI, represented by young patients with micronodular disease, usually in the lung; a second group whose metatsases can be stabilized but not cured after more than 600 mCi as cumulative doses and a third group including older patients with macronodular disease, partuclarly in the bones, who do not responde to RAI and progress rapidly to the exitus. It is apparent from this study that, continuing RAI therapy after 600 mCi usually has no benefit.
FOLLOW-UP OF CANCER PATIENTS: THE SERUM TG ASSAY
After initial ablation patients are given TSH-suppressive doses of l-thyroxine. This therapy as dual aims: to replace the thyroid hormone function and to inhibit the growth of potential residual disease..Two-three months later serum TSH, free thyroid hormones and Tg concentrations are measured during l-thyroxine treatment. The results of these tests will disclose whether the l-thyroxine dose is adequate in suppressing TSH levels without inducing hyperthyroidism, but will give little information on whether the patient is in remission.
To this purpose, the most informative follow-up period is at 6-12 minths after initial treatment, when the ablative dose of radioiodine should have exerted its effect. At this point, the patients have been already attributed a risk estimate based on the results of the post-ablative WBS and serum Tg measurement (397,398). This information is important at the moment of the new evaluation. If uptake was seen outside the thyroid bed on the post-therapy WBS, the patient must be considered in high risk of recurrence or persistent disease, while if no uptake was seen outside the thyroid bed on the 131-I post-therapy scan, the Patient is considered at low-risk. The 6-12 month control, consists of careful neck ultrasound to detect the lymph node status and serum Tg levels (with negative anti-Tg) after stimulation with exogenous (better) or endogenous TSH. These tests are considered by many authors as sufficient to confirm complete remission (negative US and undetectable stimulated Tg). Other advocate the usefulness of a diagnostic WBS with radioiodine, aimed to ensure that thyroid ablation has been successful and to search for foci of 131-I uptake outside the thyroid bed. However, two independent studies (357,399) have shown that this diagnostic WBS are almost always negative (and sometimes false negative) thus adding very little information to that given by neck US and serum Tg measurement. Indeed, the result of serum Tg measurement is the more sensitive predictor of complete remission or persistent disease (provided that anti-Tg autoantibodies are negative) (400-404). Nearly all patients with local or distant disease have detectable or elevated serum Tg levels, while patients in stable remission have undetectable serum Tg concentrations. Compared to serum Tg measurement, the yield of the diagnostic 131-I WBS is lower. A significant proportion of patients may have elevation of serum Tg in the presence of falsely negative diagnostic WBS. A retrospective study by Cailleaux et al. (357) has shown that when serum Tg off therapy is undetectable, routine diagnostic WBS does not add any further in¬formation on the clinical status of the patient. Similar results have been obtained at the Department of Endocrinology, University of Pisa, (399) in a retrospective series of 315 patients who had undetectable serum Tg off l-thyroxine at the time of the first control after thyroid ablation. None of these patients had evidence of disease activity at WBS, and 99.4% were in complete and stable remission after 12 years of follow-up. Only two patients (0.6%) had recurrence of lymph node metastases which were treated with radioiodine therapy. Based on these studies it is possible that in the future the need for 131-I scanning may be dictated by the results of serum TG during hypothyroidism or by rhTSH-stimulation (342).
After this follow up, low-risk patients (those with an undetectable stimulated serum Tg, negative neck US and negative WBS, when performed) are considered as cured and may be followed with periodic serum Tg measurement during l-thyroxine therapy. Thyroxine therapy may be decreased to maintain a low but not suppressed serum TSH concentration (0.1-0.4 µU/ml). The risk of recurrence is in fact so low in these patients (representing more than 80% of the total) that overdosage of l-thyroxine is unjustified.
As noted the problem of antibody interference in the TG assay makes this test unreliable in 10-15% of patients. However another aspect of the antibodies should be remembered. If patients are free of thyroid cancer and the thyroid has been ablated, it appears that the antigenic stimulation necessary to maintain an anti-TG titer is gradually lost, and these antibodies disappear with a 3-6 year half-life (302). Thus, the level of anti.Tg antibodies may be used as a surrogate marker of disease. Other approaches, including the search of Tg mRNA in the blood, have been proposed, but none has entered clinical practice. Fugazzola et al (405) point out that the combination of TG RIA and TG mRNA assay offer better positive and negative predictive value than TG alone. In some studies TGmRNA analysis has proven much less reliable than serum TG assay (406).
In high risk patients, even if considered cured, suppressive doses of l-thyroxine may be continued for some years, because the risk of relapse is greater. Pujol et al evaluated a series of patients over an average of 95 months and compared those who had TSH values constantly under 0.05 mU/l to those who had all TSH values greater than 1mU/l. A lesser degree of TSH suppression was associated with an increased incidence of relapse, with a shorter average relapse-free survival (407). This observation was not sustained in another study (408). The objective of suppressive therapy in these patients should be to attain a serum TSH level of 0.1 µU/ml or less with normal free T3. In this situation, side effects such as osteoporosis, are not observed (409). Clinical and biochemical evaluation is performed annually. If serum Tg becomes detectable during follow-up, the patient should be evaluated for the presence of disease by neck us, first, and by other imaging if neck ultrasound is negative. Some authors prefer to avoid this procedure and give directly a therapeutic dose of 131-I followed by a post-therapy scan. In the absence of uptake after therapeutic doses of 131-I, any further administration of 131-I is not justified, and the site of Tg production should be searched for by other imaging techniques. If 131-I thyroid ablation has not been performed or if the patient has undergone only partial thyroid surgery (subtotal or lobectomy), follow-up should consist of clinical and ultrasound examination and serum Tg measurement. However, in this case, the sensitivity and specificity of serum Tg assay is reduced. In such patients, ant suspicious of persistent or recurrence disease should prompt to complete the initial treatment with completion thyroidectomy and/or radioiodine ablation.
During follow-up, patients may develop isolated metastases that can be approached surgically. Osseous metastases, especially from follicular cancer, may require radiotherapy or operative procedures for stabilization. Progressive growth of soft tissue or osseous metastases that are not amenable to thyroid hormone, 131I therapy, or radiotherapy should lead to consideration of systemic therapies.
UNDIFFERENTIATED CARCINOMAS
Undifferentiated carcinomas, if operable, may be treated by thyroidectomy and resection of all involved tissue in the neck. 5-10% can apparently be completely resected, and the patient can have a discrete survival, although they are never cured. Prophylactic X-radiation (5,000 rads) should be given postoperatively in all cases.. If the lesion is obviously inoperable, X-ray therapy should be the initial treatment. In some instances, the rapidly growing tumors melt almost completely for a time after X-ray therapy, giving the pa¬tient a gratifying respite from the disease.
Some anaplastic tumors have been treated with alkylating agents, and rarely, a rapidly proliferating and highly undifferentiated tumor has responded with a temporary regression. Chemotherapy is discussed below. Kim and Leeper (410) reported partial results combining low dose adriamycin (10 mg/m2) and hyperfractionated radiotherapy (160 rads twice daily) to 5760 rads. Time of survival was prolonged and local recurrences largely prevented. There has been no confirmation of this study, unfortunately.
A prospective protocol combining surgery, chemotherapy (CT),and hyperfractionated accelerated radiotherapy (RT) was employed in anaplastic thyroid carcinoma. The main toxicity was hematologic. High long-term survival was obtained when RT-CT was given after complete surgery. This protocol avoided local tumor progression, and death was mainly caused by distant metastases (411). The clinical behaviour and therapy of anaplastic carcinoma have recently been reviewed by K. Ain (412).
Despite any combination of therapies, anaplastic thyroid cancer remains one of the most aggressive human cancer, with 80% of the patients dying within 2 6 months and the remaining within 2 years. Novel treatment regimens are badly needed for this tumor.
Lymphomas
Once diagnosed, patients should be staged by chest X-ray, chest and abdominal CAT scans, bone marrow biopsy, and gallium scan. In 20-30% of patients, the lymphoma will be confined within the thyroid gland. Lymph nodes are involved in approximately 60% of the patients, and perithyroidal invasion is present in about half. When patients have undergone appropriate staging procedures, 20-30% will have Stage IIIE (lymphoma present in nodes below the diaphragm) or Stage IVE (distant extranodal metastases) disease. The selection of appropriate therapy requires prior staging of the patient. As these tumors are radiosensitive, external radiation therapy is a satisfactory treatment for Stage I-E (local disease only, no nodal involvement) and has been used for Stage II-E (nodal involvement above the diaphragm only) disease. The dose to the neck is usually 4,000 rads (40 Gy) over 4-5 weeks (248). The radiation port should include the mediastinum even in the absence of clinical involvement. When evaluating the long-term survival of patients with Stage I and Stage II disease, Souhami et al. found that of seven patients treated with radiation to the neck only, five died within 5 years, whereas in five patients receiving radiation to the neck and mediastinum, there were no deaths (249). Patients with unfavorable prognostic fac¬tors such as age >60yr, large mass or tumor necrosis, may be given chemo-therapy after radiotherapy. In radiation-treated patients with local extension or with malignant nodes, the survival after surgery is unaffected by the extent of surgery even when surgery was limited to a biopsy. With obvious extension or nodal involvement, surgery should be limited to obtaining an adequate diagnostic specimen since an attempt at complete excision may damage surrounding structures without improving survival. There is a suggestion that total thyroidectomy may improve the prognosis in patients with intrathyroidal disease only (240).
There is a growing tendency to use combination chemotherapy as the initial definitive treatment for Stage IE and IIE thyroid lymphoma, rather than radiation.. Programs may combine cyclophosphamide, adriamycin, vincristine, and prednisolone ("CHOP"). This approach may increase cure above the 30-50% found with radiotherapy alone (413). Currently most patients with Stage II-E, and patients with Stage III-E or Stage IV-E disease, as well as those who relapse after radiation therapy, are treated with chemotherapy, given every 3-4 weeks for 3-6 cycles, prior to radiation therapy if combined with that treatment. Although previous series report overall 5-year survivals of about 50%, certain subgroups have a more favorable prognosis. With appro¬priate staging to exclude Stage III-E and Stage IV-E patients, the Stanford group has reported a 3-year survival of 83% and at 3 years 75% of their StageI-E and Stage II-E patients had no evidence of disease (414). Matsuzuka et al (415) used both radiation therapy and six courses of "CHOP" chemotherapy, and report 100% survival for 8 years in a group of patients who received this treatment.
RADIATION THERAPY
The general indications for radiation therapy are given in Table 18-11. Other sources should be reviewed for details of the port, dose, and methods of irradiation.
Studies by Tubiana et al (416), Simpson (283), and Riccabona (417) have clearly established the efficacy of radiotherapy in all types of thyroid cancer. Although its therapeutic use is now well accepted, its prophylactic use (e.g., in papillary or follicular lesions with possible residual disease, or in papillary or follicular Stage III lesions after RAI ablation) is controversial. Currently, we would first treat all papillary or follicular invasive or possibly metastatic tumors with RAI. In patients under age 55, it is not clear that x-ray therapy should be added. Over age 55, 131I therapy should be followed by x-ray treatment for Stage III or potentially Stage III lesions, and any recurrence or metastasis not responding to 131I may also be treated (418).
The efficacy of external radiation therapy in the management of thyroid malignancy has been reviewed by Brierley et al, Lin et al, Farahati et al, and Tsang et al. (419-422). Brierley, recommends X-ray therapy in addition to 131I therapy in patients with papillary or follicular cancer who have probable or definitive microscopic residual disease. They gave 30 to 50 Gy over four weeks with the spinal cord dose less than 80% of the total. Radiotherapy was also advised for treatment of anaplastic thyroid carcinoma, in an attempt to achieve local control, using hyperfractionation, possibly with the addition of doxorubicin. Radiotherapy was used for treatment of lymphomas, with a total dose of 35 – 40 Gy given over four weeks, and was typically preceded by the use of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) delivered every three to four weeks in three to six cycles prior to the radiotherapy. Cause-specific survival was 82% inpatients who received combined therapy and 63% in those who got X-ray alone. Combined therapies were given to all patients except those with small bulk disease under 3 cm in size confined to the thyroid, who received radiation alone. Lin et al. (420) evaluated 699 patients with differentiated thyroid cancer who received external radiotherapy after surgery, compared to 172 who did not, and found no evidence that radiotherapy improved survival rate of patients with well differentiated thyroid cancer. These were patients with either neck nodes or local invasion.There were 172 patients who received no radiotherapy versus 32 who did. The patients were in clinical stages 2 and 3 at the time of treatment. Farahati et al (421) looked at patients with parathyroidal tumor invasion and found that adjuvant external radiotherapy improved recurrence-free survival in patients over 40 years of age. Benefit was confined to patients with papillary thyroid carcinoma. Tsang et al (422) also confirmed the benefit of radiotherapy in patients with papillary tumors who have microscopic or macroscopic residual disease.
| Table 18-11. Indications for Radiation Therapy | ||
| Tumor | Stage | Treatment(15-20 MV Electrons or Co-60) |
| Papillary or Follicular | Invasive, under age 50-55 | Treat if invasive disease is thought not to be destroyed or if neck recurrence is proven present after 131-I. Dose is 4500-500 rads. |
| Invasive or possible residual, over age 50-55 | 5000 rads* to thyroid bed after RAI | |
| Recurrent, any age | 5000* rads to thyroid bed if RAI treatment is thought not to be definitive | |
| Isolated lesion in bone | 5000-6000 rads, as required for symptoms after RAI treatment | |
| Medullary | Stage III | 4000-5000* rads to thyroid bed |
| Abnormal or increasing CT | 5000* rads to mantle | |
| Recurrent tumor | 5000-6000* rads to thyroid bed | |
| Isolated metastasis | 5000-6000* rads for symptoms | |
| Lymphoma | Stage I-E, possibly II-E | Probably chemotherapy used first, 5000 rads ** to thyroid and mantle sometimes follows |
| Anaplast | All | 4500-5500 rads** to thyroid and mantle |
Note- Spinal cord dose not to exceed *3000 or ** 3500 rads.
In a recent study (423), Hurthle cell carcinoma of the thyroid gland was found to be a radiosensitive tumor. Adjuvant radiation therapy was successful in preventing recurrence in 4 of 5 patients. Salvage radiation therapy was successful in 3 of 5 patients treated with external beam radiation therapy. Palliative radiation therapy provided sustained symptomatic relief at 67% of irradiated sites. Radiation therapy may provide palliative relief from symptomatic metastases, control recurrent tumors, and prevent recurrence of advanced resected tumors.
The indications for MTC have been described above. Stage I-E lymphomas may be treated by chemotherapy or radiotherapy. Anaplastic lesions are currently given radiotherapy after operation. It is possible that chemotherapy should be used instead of or in addition to radiotherapy in these lesions, but studies are needed to establish this point.
The exact dose must be individually determined, but usually the maximal dose is 5,000-6,000 rads, using ortho-or megavoltage, and a fractionated technique over several weeks. Dosage must be planned to assure that the spinal cord receives less than 3500 rads in order to avoid myelopathy.
SYSTEMIC THERAPY FOR METASTATIC DISEASE
Sporadic experience indicates that bleomycin (424,425), adriamycin (426), vinblastine (427), methotrexate (426), cisplatinum and other agents (428) may have value in treating disseminated thyroid tumors. However, none of these drug has given more than palliation. With the advent of new molecules targeting specific metabolic pathways involve in the generation and maintainance of thyroid cancer growth, the so call “tyrosine kinase inhibitor’, chemotherapy has been almost universally abandoned. Recent ATA guidelines (39) states that whenever systemic therapy is needed, the patient should be offered the possibility to be entered in clinical trials using these new molecules. Metastatic papillary or follicular tumors refractory to conventional therapy grow slowly and systemic therapy is not indicated until the tumor is clearly growing progressively despite hormone suppression. The same apply to the treatment of MTC (264).
RE-DIFFERENTIATION’ THERAPY
RETINOIC ACID- Experimental data from in vitro studies suggest that retinoic acid can inducere-differentiation of thyroid cancer cells with regain of iodide concentrating ability. Simon et al (429) studied 28 patients with differentiated thyroid cancer using 1.5 mg/kg retinoic acid per day for five weeks. Iodide uptake increased in eight of the patients and thyroglobulin increased in 63%. Thus retinoid appeared to reinduce iodide uptake in half of the treated patients by redifferentiation. They gave 13-cis-retinoic acid. Side effects occurred in half of the patients but were generally well tolerated. A common side effect was dryness of the skin and mucosal surfaces. The reinduction of 131I uptake allowed radioiodine therapy in several of the patients. The effect on tumor size was uncertain. Retinoic acid redifferentiation therapy has recently been reviewed by Schmutzler and Kohrle (430). They note that, of twenty documented retinoic acid treated patients, at least eight had exhibited a decrease or stabilization in tumor size, and in serum TG levels, in addition to enhanced radioiodide transport. However, this relatively positive review seems enthusiastic, considering the small evidence of improvement observed in most cases. Clearly the ben¬efits of this experimental treatment are as yet uncertain, but it is nevertheless an exciting observation that retinoic acid can have an apparent beneficial redifferentiating effect on some thyroid tumors. Histone deactylase inhibitors also have been found to restore in part the function of NIS, TPO and TG in thyroid tumor cells in vitro (431), and valproic acid also has an action to restore NIS function, probably via the same mechanism (432).
PPARgamma agonists-In vitro studies show that PPAR gamma agonists can slow thyroid tumor cell growth and induce apoptosis (433). Results of clinical trials are awaited.
ONYX-015is an E1B deleted adenovirus that replicates in cells with impaired p53 function. p53 is commonly inactivated in anaplastic thyroid cancers. In vitro studies demonstrate that this virus induced cell death in in vitro trials in anaplastic cancer cell lines, and synergized with treatment with doxorubicin and pacitaxel (434).
Adenoviral vectors producing tk, IL-2, IL-12, and GM-CSF in a cell specific manner are currently under study in animals with encouraging results. Studies in humans are so far very limited, but the methods appear to be safe and effective, especially with the immunomodulator IL-12 (434,435).
A variety of ideas on possible treatments for thyroid cancer have been reviewed by Braga-Basaria and Ringel (436).
TYROSINE KINASE INHIBITORS
Several oncogenes involved in thyroid cancer progression are tyrosine kinase receptor. Molecules that block kinase activity at distal steps in the MAP kinase pathway are logical candidate drugs for thyroid cancer. TKI being tested against differentiated thyroid cancer in clinical trials include motesanib diphosphate, axitinib, gefitinib, sorafenib and sunitinib. None of these is specific for one oncogene protein but they target several TK receptors and pro-angiogenic growth factor receptors. The results of phase II-III clinical trials conducted so far are promising with partial responses ranging 14- 32% and stable disease 50-67% (437). In addition, very recently one of this drug, vandetanib, have been approved for the treatment of progressive, metastatic MTC, based in clear evidence of beneficial effects in clinical trials (438,439). All together, the preliminary results of these trials are promising and indicate that target therapy might become the first line treatment of metastatic refractory thyroid cancer patients in the near future.
Sunitinib is a multitargeted tyrosine kinase inhibitor (TKI) that is is currently approved for the therapy of renal cell carcinoma and gastrointestinal stromal tumor. Some eficacy was found in both differentiated thyoid cancer and medullary cancer in Phase II studies, with partial response rates of 13-30%.
Sorafenib is a similar agent, also achieving significant levels of partial responses and stabilized disease. Several Phase II stuies have been reported, and a Phase III trial has been completed.
Vandetanib has been studies in Phase II trials and in a randomized, placebo-controlled phase III study in patients with both hereditary and sporadic MTC, using vandetanib 300 mg or placebo. A 54% reduction in the risk of progression was observed and also objective tumor responses in 45% of patients. The FDA approved it for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease. Motesanib, Axitinib, Pazopanib, Lenvatinib and several other related TKIs that more specifically block VEGF receptors are in Phase II or III trials.
These agents appear to block progression and in some studies decrease tumor bulk, but are not specifically cytotoxic, and tend to loose efficacy over months, presumably by mutational events in the tumor. Future studies will surely investigate serial or combined treatments, and use with other possibly cytotoxic agents.
Progress in the last few years has reinvigerated therapy for these difficult tumors, but comes with a price. Since TRK Iinhibitors affect many sites in the body, side effects are very common and often serious, but generally tolerable The major side-effects include hypertension in 20-50% of cases, diarrhea in 37-77%, fatigue in 40-50%, weight loss in 20-60%, frequent nausea, skin sensitivity, and very frequent rashes, depending on the study, dosage,and drug (439). Some of the agents induce hypothyroidism during therapy.
Most reports available are the result of clinical investigations, often by consortia made up of physicians specializing in therapy of thyroid cancer, and patients are generally advised to enter such a trial if a newer agent is used.. However these agents gradually spread into more generaized usage, by therapists who may not have extensive experience. The MD Anderson group has published suggested institutional guidelines to help provide safeguards for non-investigative use of TKIs(440).
Presentation of this chapter is supported in part by Genzyme, the makers of rhTSH (THYROGEN)
2.Namba, H, Matsuo, K, Fagin, J 1990 Clonal composition of benign and malignant human thyroid tumors. J Clin In 86:.
3.. Parma, J, Duprez, L, Van Sande, J, et al. 1993. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365:649-651
4.. Tonacchera, M, Chiovato, L, Pinchera, A, et al. 1998 Hyperfunctioning thyroid nodules in toxic multinodular goiter share activating thyrotropin receptor mutations with solitary toxic adenoma. J Clin Endocrinol Metab 83:492-8
5. Coclet, J, Foureau, F, Ketelbant, P, Galand, P, Dumont, J 1989. Cell population kinetics in dog and human adult thyroid. Clinical Endocrinol 31:655-665
6. Viglietto, G, Chiappetta, G, Martinez-Tello, F J, et al. 1995 RET/PTC oncogene activation is an early event in thyroid carcinogenesis. Oncogene 11:1207-10
7. Hamburger, J I 1980; Evolution of toxicity in solitary nontoxic autonomously functioning thyroid nodules. J Clin Endocrinol Metab 50:1089-1093
8. Silverstein, G, Burke, G, Cogan, R 1967. The natural history of the auto-nomous hyperfunctioning thyroid nodule. Ann Intern Med 67:539
9. Evered, D, Clark, F, Peterson, V 1974. Thyroid function in euthyroid subjects with autonomous thyroid nodules. Clin Endocrinol 3:149
10. Horst, W, Rosler, H, Schneider, C, Labhart, A 1967. 306 cases of toxic adenoma. Clinical aspects; findings in radioiodine diagnostics; radiochromatography and histology; results of 131I and surgical treatment. J Nucl Med 8:515
11.Parma, J, Duprez, L, Van Sande, J, et al. 1993. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365:649-651
12.Suarez, H, du Villard, J, Caillou, B, Schlumberger, M, Parmentier, C, Monier, M 1991. Gsp mutations in human thyroid tumors. Oncogene 6:677-679
13. see 10
13a. DeGroot, L 1970. Lack of iodide trapping in "cold" thyroid nodules. Acta Endocrinol Panam 1:27
14. Field, J, Larsen, P, Yamashita, K, Mashiter, K, Dekke, A 1973. Demonstration of iodide transport defect but normal iodide organification in nonfunctioning nodules of human thyroid glands. J Clin Invest 52:2404
15. Fragu, P, Nataf, B 1977. Human thyroid peroxidase activity in benign and malign thyroid disorders. J Clin Endocrinol Metab 45:1089
16. Demeester-Mirkine, N, Van Sande, J, Corvilain, H, Dumont, J 1975. Benign thyroid nodule with normal iodide trap and defective organification. J Clin Endocrinol Metab 41:1169
17. Burke, G, Szabo, M 1972. Dissociation of in vivo and in vitro "autonomy" in hyperfunctioning thyroid nodules. J Clin Endocrinol Metab 35:199
18. Sande Van, J, Mockel, J, Boeynaems, J, Dor, P, Andry, G, Dumont, J 1980. Regulation of cyclic nucleotide and prostaglandin formation in normal human thyroid tissue and in autonomous nodules. J Clin Endocrinol Metab 50:776
19. Smanik, P, Ryu, K-Y, Thel, K, Mazzaferri, E, Jhiang, S 1997. Expression; exon-intron organization; and chromosome mapping of the human sodium iodide symporter. Endocrinology 138:3555-8
20.Arturi, F, Russo, D, Schlumberger, M, et al. 1998. Iodine symporter gene expression in human thyroid tumors. J Clin Endocrinol Metab 83:2493-96
21. Thomas, C J, Buckwalter, J, Staab, E, Kerr, C 1976. Evaluation of dominant thyroid masses. Ann Surg 183:464
22. Sipple, J H 1984 Multiple endocrine neoplasia type 2 syndromes: historical perspectives. Henry Ford Hosp Med J 32:219-21
23.Schimke, R, Hartmann, W, Prout, T, Rimoin, D 1968. Syndrome of bilateral pheochromocytoma; medullary thyroid carcinoma; and multiple neuromas. N Engl J Med 279:1
24.Sapira, J, Altman, M, Vandyk, K, Shapiro, A 1965. Bilateral adrenal pheo-chromocytoma and medullary thyroid carcinoma. N Engl J Med 273:140
25. Duffy, B J, Fitzgerald, P 1950. Cancer of the thyroid in children. A report of twenty-eight cases. J Clin Endocrinol 10:1296
26. Clark, D 1955. Association of irradiation with cancer of the thyroid in children and adolescents. JAMA 159:1007
27. Modan, B, Ron, E, Werner, A 1977. Thyroid cancer following scalp irradiation. Therapeutic Radiology 123:741
28 DeGroot, L, Frohman, L, Kaplan, E, Refetoff, S e 1977. Radiation-Associated Thyroid Carcinoma. New York Grune & Stratton:539 pages
29. Refetoff, S, Harrison, J, Karanfilski, B, Kaplan, E, DeGroot, L, Bekerman, C 1975. Continuing occurrence of thyroid carcinoma after irradiation to the neck in infancy and childhood. N Engl J Med 292:171
30 DeGroot, L, Paloyan, E 1973. Thyroid carcinoma and radiation. A Chicago endemic. JAMA 225:487
31. Sokal, J 1959. The problem of malignancy in nodular goiter -- recapitulation and a challenge. JAMA 170:61
32. Veith, F, Brooks, J, Grigsby, W, Selenkow, H 1964. The nodular thyroid gland and cancer. N Engl J Med 270:431
32.1 Frates MC, Benson CB, Doubilet PM, Kunreuther E, Contreras M, Cibas ES, Orcutt J, Moore FD Jr, Larsen PR, Marqusee E, Alexander EK. Prevalence and distribution of carcinoma in patients with solitary and multiple thyroid nodules on sonography. J Clin Endocrinol Metab. 2006 Sep;91(9):3411-7.
33. Hoffman, G, Thompson, N, Heffron, C 1972. The solitary thyroid nodule. Arch Surg 105:379
33.1 Haymart MR, Repplinger DJ, Leverson GE, Elson DF, Sippel RS, Jaume JC, Chen H.Higher serum thyroid stimulating hormone level in thyroid nodule patients is associated with greater risks of differentiated thyroid cancer and advanced tumor stage.J Clin Endocrinol Metab. 2008 Mar;93(3):809-14.
34. Pacini, F, Fontanelli, M, Fugazzola, L, et al. 1994. Routine measurement of serum calcitonin in nodular thyroid diseases allows the preoperative diagnosisof unsuspected sporadic medullary thyroid carcinoma. J Clin Endocrinol Mertab 78:826-9
35. Rieu, M, Lame, M, Richard, A, et al. 1995. Prevalence of sporadic medullary thyroid carcinoma. the importance of routine measurement of serum calcitonin in the diagnostic evaluation of thyroid nodules. Clin Endocrinol (Oxf) 42:453-7
36. Niccoli, P, Wion-Barbot, N, Caron, P, et al. 1997. Interest of routine measurement of serum calcitonin. Study in a large series of thyroidectomized patients. J Clin Endocrinol Metab 82:338-341
37. Gagel, R 1997. The goitrous patient with an elevated serum calcitonin -- what to do? J Clin Endocrinol Metab 82:335
38. Hamlin, E, Vickery, A 1956. Needle biopsy of the thyroid gland. N Engl J Med 254:742
39. Walfish, P, Hazani, E, Strawbridge, H, Miskin, M, Rosen, B 1977. Combined ultrasound and needle aspiration cytology in the assessment and management of hypofunctioning thyroid nodule. Ann Intern Med 87:270
40. Gershengorn, M, McClung, M, Chu, W, Hanson, T, Weintraub, B, Robbins, J 1977. Fine-needle aspiration cytology in the preoperative diagnosis of thyroid nodules. Ann Intern Med 87:265
41.Baloch, Z, Sack, M, Livolsi, V, Gupta, P 1998. Fine-needle aspiration of thyroid. an institutional experience. Thyroid 8:565-9
42. Benvenga, S, Bartolone, L, Squadrito, S, Trimarchi, F 1997. Thyroid hormone autoantibodies elicited by diagnostic fine needle biopsy. J Clin Endocrinol Metab 82:4217-4223
43 Wilems, J-S, Lowhagen, T 1981. Fine needle aspiration cytology in thyroid disease. . Clin Endocrinol Metab 2:247
44 Gharib, H, Goellner, J, Johnson, D 1993. Fine needle aspiration cytology of the thyroid. A 12-year experience with 11;000 biopsies. Clin Lab Med 13:699-709
44.1 Tee YY, Lowe AJ, Brand CA, Judson RT.Fine-needle aspiration may miss a third of all malignancy in palpable thyroid nodules: a comprehensive literature review.Ann Surg. 2007 Nov;246(5):714-20.
45. Pacini, F, Fugazzola, L, Lippi, F, et al. 1992 Detection of thyroglobulin in fine needle aspirates of nonthyroidal neck masses. a clue to the diagnosis of metastatic thyroid cancer. J Clin Endocrinol Metab 74:1401-4
46. Chow, L S, Gharib, H, Goellner, J R, van Heerden, J A 2001 Nondiagnostic thyroid fine-needle aspiration cytology: management dilemmas. Thyroid 11:1147-51
47. Khurana, K, Richards, V, Chopra, P, Izquierdo, R, Rubens, D, Mesonero, C 1998. The role of ultrasonography-guided fine-needle aspiration biopsy in the management of nonpalpable and palpable thyroid nodules. Thyroid 8:511
48. Erdogan, M, Kamel, N, Aras, D, Akdogan, A, Baskal, N, Erdogan, G 1998. Value of reaspirations in benign nodular thyroid disease. Thyroid 8:1087
49. Bieche, I, Ruffet, E, Zweibaum, A, Vilde, F, Lidereau, R, Franc, B 1997. MUC1 Mucin gene; transcripts; and protein in adenomas and papillary carcinomas of the thyroid. Thyroid 7:725
50 Brousset, P, Chaouche, N, Leprat, F, et al. 1997. Telomerase activity in human thyroid carcinomas originating from the follicular cells. J Clin Endocrinol Metab 82:4214-4216
51 Khoo, M L, Beasley, N J, Ezzat, S, Freeman, J L, Asa, S L 2002 Overexpression of cyclin D1 and underexpression of p27 predict lymph node metastases in papillary thyroid carcinoma. J Clin Endocrinol Metab 87:1814-8
51a. Maruta, J; Hashimoto, H; Yamashita, H; Yamashita, H; Noguchi, S. Diagnostic applicability of dipeptidyl aminopeptidase IV activity in cytological samples for differentiating follicular thyroid carcinoma from follicular adenoma. Arch Surg 139 83-88 2004.
51b. Durand S, Ferraro-Peyret C, Selmi-Ruby S, Paulin C, El Atifi M, Berger F, Berger-Dutrieux N, Decaussin M, Peix JL, Bournaud C, Orgiazzi J, Borson-Chazot F, Rousset B Evaluation of gene expression profiles in thyroid nodule biopsy material to diagnose thyroid cancer. J Clin Endocrinol Metab. 2008 Apr;93(4):1195-202.
51c Shibru D, Hwang J, Khanafshar E, Duh QY, Clark OH, Kebebew E.Does the 3-gene diagnostic assay accurately distinguish benign from malignant thyroid neoplasms?Cancer. 2008 Sep 1;113(5):930-5.
52 Clark, O, Okerlund, M, Cavalieri, R, Greenspan, F 1979. Diagnosis and treatment of thyroid; parathyroid; and thyroglossal duct cysts. J Clin Endocrinol Metab 48:983
52.1 Leboulleux S, Girard E, Rose M, Travagli JP, Sabbah N, Caillou B, Hartl DM, Lassau N, Baudin E, Schlumberger M Ultrasound criteria of malignancy for cervical lymph nodes in patients followed up for differentiated thyroid cancer. J Clin Endocrinol Metab. 2007 Sep;92(9):3590-4
53. Marcocci, C, Vitti, P, Cetani, F, Catalano, F, Concetti, R, Pinchera, A 1991. Thyroid ultrasonography helps to identify patients with diffuse lymphocytic thyroiditis who are prone to develop hypothyroidism. J Clin Endocrinol Metab 72:209-13
54. Kendall, L, Condon, R 1969. Prediction of malignancy in solitary thyroid nodules. Lancet 1:1071
55. Miller, J, Hamburger, J 1965.. The thyroid scintigram. 1. The hot nodule. Radiology 84:66
56. Attie, J 1960. The use of radioactive iodine in the evaluation of thyroid nodules. Surgery 47:611
57. Dische, S 1964. The radioisotope scan applied to the detection of carcinoma in thyroid swellings. Cancer 17:473
58. Fujimoto, Y, Oka, A, Nagataki, S 1972. Occurrence of papillary carcinoma in hyperfunctioning thyroid nodule. Report of a case. Endocrinol Jpn 19:371
59. Scott, M, Crawford, J 1976. Solitary thyroid nodules in childhood. Is the incidence of thyroid carcinoma declining? Pediatrics 58:521
60.Messaris, G, Evangelou, G, Tountas, C 1973. Incidence of carcinoma in cold nodules of the thyroid gland. Surgery 74:447
61. Van den Bruel, A, Maes, A, De Potter, T, et al. 2002 Clinical relevance of thyroid fluorodeoxyglucose-whole body positron emission tomography incidentaloma. J Clin Endocrinol Metab 87:1517-20
62. Hakama, M Berlin; Springer-Verlag Different world thyroid cancer rates; in Hedinger CE (ed). Thyroid Cancer. International Union Against Cancer Monograph Series:Vol 12.
63. Young, J J, Percy, C, Asire, A e 1082 pages Surveillance; Epidemiology; and End Results. Incidence and Mortality Data; 1973-77. National Cancer Institute Monograph 57 NIH Publication No. 81:2330
64. Goldstein, R, Hart, I 1983. Follow-up of solitary autonomous thyroid nodules treated with 131I. New Engl J Med 309:1473
65. Gorman, C, Robertson, J 1978. Radiation dose in the selection of 131I or surgical treatment for toxic thyroid adenoma. Ann Intern Med 89:85
66. Paracchi, A, Ferrari, C, Livraghi, T, et al. 1992. Percutaneous intranodular ethanol injection. A new treatment for autonomous thyroid adenoma. J Endocrinol Invest 15:353-362
67. Lippi, F, Ferrari, C, Manetti, L, et al. 1996. Treatment of solitary autonomous thyroid nodules by percutaneous ethanol injection. results of an Italian multicenter study. The Multicenter Study Group. J Clin Endocrinol Metab 81:3261-3264
68. Monzani, F, Caraccio, N, Goletti, O, et al. S54-8 Treatment of hyperfunctioning thyroid nodules with percutaneous ethanol injection. Eight years' experience. Exp Clin Endocrinol Diabetes 106: Suppl 4
69. Hamburger, J I 1980; Evolution of toxicity in solitary nontoxic autonomously functioning thyroid nodules. J Clin Endocrinol Metab 50:1089-1093
70. McCowen, K, Reed, J, BL., F 1980. The role of thyroid therapy in patients with thyroid cysts. Amer J Med 68:853
71. Treece, G, Georgitis, W, Hofeldt, F 1983 Resolution of recurrent thyroid cysts with tetracycline instillation. Arch Int Med 143: 2285.
72. Monzani, F, Lippi, F, Goletti, O, et al. 1994. Percutaneous aspiration and ethanol sclero-therapy for thyroid cysts. J Clin Endocrinol Metab 78:800-802
73. Zingrillo, M, Torlontano, M, Ghiggi, M, et al. 1996. Percutaneous ethanol injection of large thyroid cystic nodules. Thyroid 6:403-8
74.Del Prete, S, Caraglia, M, Russo, D, et al. 2002 Percutaneous ethanol injection efficacy in the treatment of large symptomatic thyroid cystic nodules: ten-year follow-up of a large series. Thyroid 12:815-21
75. see 70
76. Papini, E, Bacci, V, Panunzi, C, et al. 1993. A prospective randomized trial of levothyroxine suppressive therapy for solitary thyroid nodules. Clin Endocrinol 38:507-513
77. Burch, H 1995. Evaluation and management of the solid thyroid nodule. Endocrinol Metab Clin N Am 24:663-710
78. Mainini, E, Martinelli, I, Morandi, G, Villa, S, Stefani, I, Mazzi, C 1995. Levothyroxine suppressive therapy for solitary thyroid nodule. J Endocrinol Invest 18:796-799
79. Cooper, D 1995. Clinical review 66. Thyroxine suppression therapy for benign nodular disease. J Clin Endocrinol Metab 80:331-334
80. Castro, M R, Caraballo, P J, Morris, J C 2002 Effectiveness of thyroid hormone suppressive therapy in benign solitary thyroid nodules: a meta-analysis. J Clin Endocrinol Metab 87:4154-9
81 Zelmanovits, F, Genro, S, Gross, J 1998. Suppressive therapy with levothyroxine for solitary thyroid nodules. A double-blind controlled clinical study and cumulative meta-analyses. J Clin Endocrinol Metab 83:3881-3885
82. Herms, A, Huysmans, D 1998. Treatment of benign nodular thyroid disease. N Engl J Med 312:601-4
83. Larosa, G, Ippolito, A, Luppo, L, et al. 1996. Cold thyroid nodule reduction with l-thyroxine can be predicted by initial nodule volume and cytological characteristics. J Clin Endocrinol Metab; 81:4385-7
83.a. Sdano MT, Falciglia M, Welge JA, Steward DL. Efficacy of thyroid hormone suppression for benign thyroid nodules: meta-analysis of randomized trials Otolaryngol Head Neck Surg. 2005 Sep;133(3):391-6
84. Wemeau, J L, Caron, P, Schvartz, C, et al. 2002 Effects of thyroid-stimulating hormone suppression with levothyroxine in reducing the volume of solitary thyroid nodules and improving extranodular nonpalpable changes: a randomized, double-blind, placebo-controlled trial by the French Thyroid Research Group. J Clin Endocrinol Metab 87:4928-34
85 see 77
86 Morita, T, Tamai, H, Ohshima, A, et al. 1989. Changes in serum thyroid hormone; thyrotropin and thyroglobulin concentrations during thyroxine therapy in patients with solitary thyroid nodules. J Clin Endocrinol Metab 69:227
87 Woolner, L, Beahrs, O, Black, B, McConahey, W, Keating, F J vol 12 Long term survival rates. in Hedinger Chr E (ed). Thyroid Cancer. International Union Against Cancer: Monograph Series
88. Fogelfeld, L, Wiviott, M, Shore-Freedman, E, et al. 1989. Recurrence of thyroid nodules after surgical removal in patients irradiated in childhood for benign conditions. N Engl J Med 320:835-840
89. Bennedbaek, F, Hegedus, L 1999. Percutaneous ethanol injection therapy in benign solitary solid cold thyroid nodules. A randomized trial comparing one injection with three injections. Thyroid 9:225-233
90. Khurana, K, Labrador, E, Izquierdo, R, Mesonero, C, Pisharodi, L 1999. The role of fine-needle aspiration biopsy in the management of thyroid nodules in children; adolescents; and young adults. A multi-institutional study. Thyroid 9:383
91.Tan, G, Gharib, H 1997. Thyroid incidentalomas. management approaches to nonpalpable nodules discovered incidentally on thyroid imaging. Ann Intern Med 126:226-31
92. Franceschi, S, Boyle, P, Maissonneuve, P, et al. 1993. The epidemiology of thyroid carcinoma. Critical Reviews in Oncogenesis 4:25
93. Parkin, D, Muir, C, Whelan, S, Gao, Y, Fenlay, J, Powell, J Vol. 6 Cancer incidence in five continents. IARC Scientific Publication 120 International Agency for Research on Cancer: Lyon
94 Goodman, M, Yoshizawa, C, Kolonel, L 1988. Descriptive epidemiology of thyroid cancer in Hawaii. Cancer 61:1272
95. Spitz, M, Sider, J, Katz, R, Pollack, E, Newell, G 1988. Ethnic patterns of thyroid cancer incidence in the United States; 1973-1981. Int J Cancer 42:549
95.1. .Davies L, Welch HG Increasing incidence of thyroid cancer in the United States, 1973-2002. JAMA. 2006 May 10;295(18):2164-7.
95.2. Enewold L, Zhu K, Ron E, Marrogi AJ, Stojadinovic A, Peoples GE, Devesa SS.Rising thyroid cancer incidence in the United States by demographic and tumor characteristics, 1980-2005.Cancer Epidemiol Biomarkers Prev. 2009 Mar;18(3):784-91
96. Mortensen, J, Woolner, L, Bennett, W 1955. Gross and microscopic findings in clinically normal thyroid glands. J Clin Endocrinol Metab 15:1270
97. Silverberg, S, Vidone, R 1966. Metastatic tumors in the thyroid. Pac Med & Surg 74:175
98. Hundahl, S A, Fleming, I D, Fremgen, A M, Menck, H R 1998 A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995 [see commetns]. Cancer 83:2638-48
99. Wahner, H, Cuello, C, Correa, P, Uribe, L, Gaitan, E 1966. Thyroid carcinoma in an endemic goiter area; Cali; Colombia. Am J Med 40:58
100. Belfiore, La Rosa, G, La Porta, G, et al. 1992. Cancer risk in patients with cold thyroid nodules. relevance of iodine intake; sex; age; and multinodularity. Am J Med 93:363
100.1. Sandeep TC, Strachan MW, Reynolds RM, Brewster DH, Scelo G, Pukkala E, Hemminki K, Anderson A, Tracey E, Friis S, McBride ML, Kee-Seng C, Pompe-Kirn V, Kliewer EV, Tonita JM, Jonasson JG, Martos C, Boffetta P, Brennan P. Second primary cancers in thyroid cancer patients: a multinational record linkage study.J Clin Endocrinol Metab. 2006 May;91(5):1819-25
101. Namba, H, Matsuo, K, Fagin, J 1990 Clonal composition of benign and malignant human thyroid tumors. J Clin In 86:.
102. Coclet, J, Foureau, F, Ketelbant, P, Galand, P, Dumont, J 1989. Cell population kinetics in dog and human adult thyroid. Clinical Endocrinol 31:655-665
103. Curran, P, DeGroot, L 1991. The effect of hepatic enzyme-inducing drugs on thyroid hormones and the thyroid gland. Endocrine Rev 12:135-150
104. Namba, H, Gutman, R, Matsuo, K, Alvarez, A, Fagin, J 1990 H-Ras protooncogene mutations in human thyroid neoplasms. J Clin Endocrinol Metab 71: .
105. Namba, H, Rubin, S, Fagin, J 1990. Point mutations of Ras oncogenes are an early event in thyroid tumorigenesis. Molecul Endocrinol 4:1474
106. Franceschi, S, Boyle, P, Maissonneuve, P, et al. 1993. The epidemiology of thyroid carcinoma. Critical Reviews in Oncogenesis 4:25.
107. Parkin, D, Muir, C, Whelan, S, Gao, Y, Fenlay, J, Powell, J Vol. 6 Cancer incidence in five continents. IARC Scientific Publication 120 International Agency for Research on Cancer: Lyon.
108. Goodman, M, Yoshizawa, C, Kolonel, L 1988. Descriptive epidemiology of thyroid cancer in Hawaii. Cancer 61:1272.
109. Spitz, M, Sider, J, Katz, R, Pollack, E, Newell, G 1988. Ethnic patterns of thyroid cancer incidence in the United States; 1973-1981. Int J Cancer 42:549.
110. Mortensen, J, Woolner, L, Bennett, W 1955. Gross and microscopic findings in clinically normal thyroid glands. J Clin Endocrinol Metab 15:1270.
111. Silverberg, S, Vidone, R 1966. Carcinoma of the thyroid in surgical and post-mortem material. Analysis of 300 cases at autopsy and literature review. Ann Surgery 164:291.
112. Hundahl, S A, Fleming, I D, Fremgen, A M, Menck, H R 1998 A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995 [see commetns]. Cancer 83:2638-48.
113. Wahner, H, Cuello, C, Correa, P, Uribe, L, Gaitan, E 1966. Thyroid carcinoma in an endemic goiter area; Cali; Colombia. Am J Med 40:58.
114. Belfiore, La Rosa, G, La Porta, G, et al. 1992. Cancer risk in patients with cold thyroid nodules. relevance of iodine intake; sex; age; and multinodularity. Am J Med 93:363.
115. Namba, H, Matsuo, K, Fagin, J 1990 Clonal composition of benign and malignant human thyroid tumors. J Clin In 86.
116. Coclet, J, Foureau, F, Ketelbant, P, Galand, P, Dumont, J 1989. Cell population kinetics in dog and human adult thyroid. Clinical Endocrinol 31:655-665.
117. Curran, P, DeGroot, L 1991. The effect of hepatic enzyme-inducing drugs on thyroid hormones and the thyroid gland. Endocrine Rev 12:135-150.
118. Namba, H, Gutman, R, Matsuo, K, Alvarez, A, Fagin, J 1990 H-Ras protooncogene mutations in human thyroid neoplasms. J Clin Endocrinol Metab 71.
119. Namba, H, Rubin, S, Fagin, J 1990. Point mutations of Ras oncogenes are an early event in thyroid tumorigenesis. Molecul Endocrinol 4:1474.
120. Karga, H, Lee, J-K, Vickery, A, Thor, A, Gaz, R, Jameson, J 1991. Ras oncogene mutations in benign and malignant thyroid neoplasms. J Clin Endocrinol Metab 73:832.
121. Santoro, M, Carlomagno, F, Hay, I, et al. 1992. Ret oncogene activation in human thyroid neoplasms is restricted to the papillary cancer subtype. J Clin Invest 89:1517-1522
122. Jhiang, S, Sagartz, J, Tong, Q, et al. 1996. Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology 137:375-378.
123. Nikiforova, MN; Kimura, ET; Gandhi, M; Biddinger, PW; Knauf, JA; Basolo, F; Zhu, Z; Giannini, R; Salvatore, G; Fusco, A; Santoro, M; Fagin, JA; Nikiforov, YE. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab 88 5399-5404 2003.
124. Giannini R, Ugolini C, Lupi C, Proietti A, Elisei R, Salvatore G, Berti P, Materazzi G, Miccoli P, Santoro M, Basolo F. The Heterogeneous Distribution of Distinct Tumor BRAF mutations in Multifocal Papillary J Clin Endocrinol Metab. 2007 Sep;92(9):3511-6
125. Kroff, T, Sarraf, P, Pecciarini, L, et al. 2000. PAX8-PPAR(1 fusion oncogene in human thyroid carcinoma. Science 289:1357-1360
126. Powell, JG; Allard, BL; Sahin, M; Wang, X-L; Wang, X; Hay, ID; Hiddinga, HJ; Deshpande, SS; Kroll, TG; Grebe, SKG; Eberhardt, NL; McIver, B. The PAX8/PPAR( fusion oncoprotein transforms immortalized human thyrocytes through a mechanism probably involving wild-type PPAR inhibition. Oncogene 2003.
127. Marques, A R, Espadinha, C, Catarino, A L, et al. 2002 Expression of PAX8-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J Clin Endocrinol Metab 87:3947-52
128. Vivaldi, A, Pacini, F, Martini, F, et al. 2003 Simian virus 40-like sequences from early and late regions in human thyroid tumors of different histotypes. J Clin Endocrinol Metab 88:892-9
129. Puzianowska-Kuznicka, M, Krystyniak, A, Madej, A, Cheng, S Y, Nauman, J 2002 Functionally impaired TR mutants are present in thyroid papillary cancer. J Clin Endocrinol Metab 87:1120-8
130. Takano, T; Miyauchi, A; Yoshida, H; Nakata, Y; Kuma, K; Amino, N. Expression of TR(1 mRNAs with functionally impaired mutations is rare in thyroid papillary carcinoma. J Clin Endocrinol Metab. 88:3447-3449, 2003.
131. Liu, R T, Huang, C C, You, H L, et al. 2002 Overexpression of tumor susceptibility gene TSG101 in human papillary thyroid carcinomas. Oncogene 21:4830-7.
132. Parma, J, Duprez, L, Van Sande, J, et al. 1993. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365:649-651
133. Suarez, H, du Villard, J, Caillou, B, Schlumberger, M, Parmentier, C, Monier, M 1991. Gsp mutations in human thyroid tumors. Oncogene 6:677-679
134. Matsuo, K, Friedman, E, Gejman, P, Fagin, J 1993. The thyrotropin receptor (TSH-R) is not an oncogene for thyroid tumors. Structural studies of the TSH-R and the -subunit of GS in human thyroid neoplasms. J Clin Endocrinol Metab 76:1446-1451.
135. Shi, Y, Zou, M, Farid, N 1993. Expression of thyrotrophin receptor gene in thyroid carcinoma is associated with a good prognosis. Clinical Endocrinol 39:269-274.
136. Fagin, J, Matsuo, K, Karmakar, A, Chen, D, Tang, S-H, Koeffler, H 1993. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest 91:179-184.
137. Wada, N, Duh, Q Y, Miura, D, Brunaud, L, Wong, M G, Clark, O H 2002 Chromosomal aberrations by comparative genomic hybridization in hurthle cell thyroid carcinomas are associated with tumor recurrence. J Clin Endocrinol Metab 87:4595-601.
138. Larsson, C, Skogseid, B, Oberg, K, Nakamura, Y, Nordenskjold, M 1988. Multiple endocrine neoplasia type I gene maps to chromosome 11 and is lost in insulinoma. Nature 332:85-87.
139. Quadro, L, Panariello, L, Salvatore, D, et al. 1994. Frequent RET proto-oncogene mutations in multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab 79:590-594.
140. Romei C, Elisei R, Pinchera A, Ceccherini I, Molinaro E, Mancusi F, Martino E, Romeo G, Pacini F. 1996. Somatic mutations of the ret protooncogene in sporadic medullary thyroid carcinoma are not restricted to exon 16 and are associated with tumor recurrence. J Clin Endocrinol Metab.81:1619-22.
141. Elisei R, Cosci B, Romei C, Bottici V, Renzini G, Molinaro E, Agate L, Vivaldi A, Faviana P, Basolo F, Miccoli P, Berti P, Pacini F, Pinchera A. 2008. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 93:682-7.
142. Loh, K C 1997 Familial nonmedullary thyroid carcinoma: a meta-review of case series. Thyroid 7:107-13..
143. Fagin, J 1997. Familial nonmedullary thyroid carcinoma - The case for genetic susceptibility. J Clin Endocrinol Metab 82:342-344.
144. Capezzone M, Marchisotta S, Cantara S, Busonero G, Brilli L, Pazaitou-Panayiotou K, Carli A, Caruso G, Toti P, Capitani S, Pammolli A, Pacini F. Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocr Relat Cancer 15:1075-81, 2008.
145. Marco Capezzone, Silvia Cantara, Stefania Marchisotta, Sebastiano Filetti, Maria Margherita De Santi, Benedetta Rossi, Giuseppe Ronga, Cosimo Durante, Furio Pacini. Short telomeres, hTERT gene amplification and increased telomerase activity in the blood of familial papillary thyroid cancer patients. J Clin Endocrinol Metab 93:3950-7, 2008.
146. Capezzone M, Cantara S, Marchisotta S, Busonero G, Formichi C, Benigni M, Capuano S, Toti P, Pazaitou-Panayiotou K, Caruso G, Carli AF, Palummo N, Pacini F. Telomere Length in Neoplastic and Nonneoplastic Tissues of Patients with Familial and Sporadic Papillary Thyroid Cancer. J Clin Endocrinol Metab. 2011 Aug 24. [Epub ahead of print]
147. Lloyd, K, Dennis, M 1963. Cowden's disease. A possible new symptom complex with multiple system involvement. Annals Int Med 58:136
148. Malchoff, C D, Malchoff, D M 2002 The genetics of hereditary nonmedullary thyroid carcinoma. J Clin Endocrinol Metab 87:2455-9
149. Parkin, J 1981. Familial multiple glomus tumors and pheochromocytomas. Ann Otol 90:.60
150. Camiel, M, Mule, J, Alexander, L, Benninghoff, D 1968. Association of thyroid carcinoma with Gardner's syndrome in siblings. N Engl J Med 278:1056
151. Stratakis, C, Courcoutsakis, N, Abati, A, et al. 1997. Thyroid gland abnormalities in patients with the syndrome of spotty skin pigmentation; myxomas; endocrine overactivity; and schwannomas (Carney Complex). J Clin Endocrinol Metab 82:2037-2043.
152. Malchoff, C, Sarfarazi, M, Tendler, B, et al. 2000. Papillary thyroid carcinoma associated with papillary renal neoplasia. genetic linkage analysis of a distinct heritable tumor syndrome. J Clin Endocrinol Metab 85:1758-1764.
153. Wollman, S 1963. Production and properties of transplantable tumors of the thyroid gland in the Fischer rat. Recent Prog Hormone Research 19:579.
154. Frantz, V, Kligerman, M, Harland, W, Phillips, M, Quimby, E 1957. A comparison of the carcinogenic effect of internal and external irradiation on the thyroid gland of the male Long- Evans rat. Endocrinology 61:574.
155. Doniach, I 1953. The effect of radioactive iodine alone and in combination with nethylthiouracil upon tumor production in the rat's thyroid gland. Br J Cancer 7:181.
156. Maloof, F, Dobyns, B, Vickery, A 1952. The effects of various doses of radioactive iodine on the function and structure of the thyroid of the rat. Endocrinology 50:612.
157. Dobyns, B, Didtschenko, I 1961. Nuclear changes in thyroidal epithelium following radiation from radioiodine. J Clin Endocrinol Metab 21:699
158. Duffy, B J, Fitzgerald, P 1950. Cancer of the thyroid in children. A report of twenty-eight cases. J Clin Endocrinol 10:1296.
159. Clark, D 1955. Association of irradiation with cancer of the thyroid in children and adolescents. JAMA 159:1007.
160. Modan, B, Ron, E, Werner, A 1977. Thyroid cancer following scalp irradiation. Therapeutic Radiology 123:741.
161. DeGroot, L, Frohman, L, Kaplan, E, Refetoff, S e 1977. Radiation-Associated Thyroid Carcinoma. New York Grune & Stratton:539 pages.
162. Refetoff, S, Harrison, J, Karanfilski, B, Kaplan, E, DeGroot, L, Bekerman, C 1975. Continuing occurrence of thyroid carcinoma after irradiation to the neck in infancy and childhood. N Engl J Med 292:171.
163. DeGroot, L, Paloyan, E 1973. Thyroid carcinoma and radiation. A Chicago endemic. JAMA 225:487.
164. Pifer, J, Hempelmann, L, Dodge, H, Hodges, F 1968. Neoplasms in the Ann Arbor Series of thymus-irradiated children; a second survey. Am J Roent; Rad Ther & Nucl Med 103:13.
165. Saenger, E, Silverman, F, Sterling, T, Turner, M 1960. Neoplasia following therapeutic irradiation for benign conditions in childhood. Radiology 74:.889.
166. Beach, S, Dolphin, G 1962. A study of the relationship between X-ray dose delivered to the thyroids of children and the subsequent development of malignant tumors. Phys Med Biol 6:583
167. Winship, T, Rosvoll, R 1961. Thyroid carcinoma in childhood. Cancer 14:734.
168. Maxon, H, Thomas, S, Saenger, E, Buncher, C, Kereiakes, J 1977. Ionizing irradiation and the induction of clinically significant disease in the human thyroid gland. Am J Med 63:967.
169. Hollowell, JG; Staehling, NW; Flanders, WD; Hannon, WH; Gunter, EW; Spencer, CA; Braverman, LE. Serum TSH, T4, and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab 87:489-499, 2002.
170. Perkel, V, Gail, M, Lubin, J, et al. 1988. Radiation-induced thyroid neoplasms. Evidence for familial susceptibility factors. J Clin Endocrinol Metab 66:1316.
171. Hanford, J, Quinby, E, Frantz, V 1962. Cancer arising many years after radiation therapy. JAMA 181:404.
172. DeLawter, D, Winship, T 1963. Follow-up study of adults treated with Roentgen rays for thyroid disease. Cancer 16:1028.
173. Parker, L, Belsky, J, Yamamoto, T, Kawamoto, S, Keehn, R 1974. Thyroid carcinoma after exposure to atomic radiation. Ann Intern Med 80:600.
174. Hancock, S, Cox, R, McDougall, I 1991. Thyroid diseases after treatment of Hodgkin's disease. N Engl J Med 325:599.
175. Pacini, F, Vorontsova, T, Demidchik, E, et al. 1997. Post-chernobyl thyroid carcinoma in Belarus children and adolescents. Comparison with naturally occurring thyroid carcinoma in Italy and France. J Clin Endocrinol Metab 82:3563-3569.
176. Favus, J, Schneider, A, Stachura, M, et al. 1976. Thyroid cancer occurring as a late consequence of head-and-neck irradiation. N Engl J Med 294:1019.
177. Spitalnik, P, Straus, F 1978. Patterns of human thyroid parenchymal reaction following low-dose childhood irradiation. Cancer 41:1098.
178. Roudebush, C, Asteris, G, DeGroot, L 1978. Natural history of radiation-associated thyroid cancer. Arch Intern Med 138:1631.
179. Rao, D, Frame, B, Miller, M, Kleerekoper, M, Block, M, Parfitt, A 1980. Hyperparathyroidism following head and neck irradiation. Arch Intern Med 140:205.
180. Conard, R 1975. A twenty-year review of medical findings in a Marshallese population accidentally exposed to radioactive fallout. Published by Brookhaven National Laboratory (BNL No. 50424) Upton: New York.
181. Vickery, A The Williams & Wilkins Co Thyroid alterations due to irradiation. The Thyroid International Academy of Pathology Monograph No 5: Baltimore.
182. Cantolino, S, Schmickel, R, Ball, M, Cisar, C 1966. Persistent chromosomal aberrations following radioiodine therapy for thyrotoxicosis. N Engl J Med 275:739.
183. Sheline, G, Lindsay, S, McCormack, K, Galante, M 1962. Thyroid nodules occurring late after treatment of thyrotoxicosis with radioiodine. J Clin Endocrinol Metab 2: 8.
184. Dobyns, B, Sheline, G, Workman, J, Tompkins, E, McConahey, W, Becker, D 1974. Malignant and benign neoplasms of the thyroid in patients treated for hyperthyroidism. A report of the cooperative thyrotoxicosis therapy follow-up study. J Clin Endocrinol Metab 38:976.
185. Holm, LE, Dahlqvist, I, Israelsson, A, Lundell, G 1980. Malignant thyroid tumors after iodine 131-therapy. N Engl J Med 303:188.
186. Holm, L-E, Eklund, G, Lundell, G 1980. Incidence of malignant thyroid tumors in humans after exposure to diagnostic doses of iodine-131. II. Estimation of thyroid gland size; thyroid radiation; and predicted versus observed number of malignant thyroid tumors. J Natl Cancer Inst 65:1221.
187. Stanbury, J F g p 273 in Stanbury, JB; Wyngaarden, JB; Fredrickson, DS (eds). The Metabolic Basis of Inhterited Disease. New York: McGraw-Hill Book Co Inc.
188. McGirr, E, Clement, W, Currie, A, Kennedy, J 1959. Impaired dehalogenase activity as a cause of goiter with malignant changes. Scott Med J 4:232.
189. Elman, D 1958. Familial association of nerve deafness with nodular goiter and thyroid carcinoma. N Engl J Med 259:219.
190. Medeiros-Neto, G, Oliveira, N 1970. Follicular adenocarcinoma of thyroid associated with congenital hyperplastic goiter. Acta Endocrinol Panam 1:73.
191. Cooper, D, Axelrod, L, DeGroot, L, Vickery, A, Maloof, F 1981. Congenital goiter and the development of metastatic follicular carcinoma with evidence for a leak of nonhormonal iodide. Clinical; pathological; kinetic; and biochemical studies and a review of the literature. J Clin Endocrinol Metab 52:294.
192. Sloan, L 1954. Of the origin; characteristics and behaviour of thyroid cancer. J Clin Endocrinol Metab 14:1309.
193. Ellenbert, A, Goldman, L, Gordan, G, Lindsay, S 1962. Thyroid carcinoma in patients with hyperparathyroidism. Surgery 51:708.
194. LiVolsi, V, Feind, C 1976. Parathyroid adenoma and nonmedullary thyroid carcinoma. Cancer 38:1391.
195. Shapiro, S, Friedman, N, Perzik, S, Catz, B 1970. Incidence of thyroid carcinoma in Graves' disease. Cancer 26:1261
196. Fiore E, Rago T, Latrofa F, Provenzale MA, Piaggi P, Delitala A, Scutari M, Basolo F, Di Coscio G, Grasso L, Pinchera A, Vitti P.Hashimoto's thyroiditis is associated with papillary thyroid carcinoma: role of TSH and of treatment with L-thyroxine.Endocr Relat Cancer. 2011, 18:429-37.
197. Boi F, Lai ML, Marziani B, Minerba L, Faa G, Mariotti S. High prevalence of suspicious cytology in thyroid nodules associated with positive thyroid autoantibodies.Eur J Endocrinol. 2005;153:637-42.
198. Belfiore, A, Garofalo, M, Giuffrida, D, et al. 1990. Increased aggressiveness of thyroid cancer in patients with Graves' disease. J Clin Endocrinol Metab 70:830.
199. Pellegriti, G, Belfiore, A, Giuffrida, D, Lupo, L, Vigneri, R 1998. Outcome of differentiated thyroid cancer in Graves' patients. J Clin Endocrinol Metab 83:2805.
200. Filetti, S, Belfiore, A, Amir, S, et al. 1988. The role of thyroid-stimulating antibodies of Graves' disease in differentiated thyroid cancer. N Engl J Med 318:753.
201. Pacini F, Elisei R, Di Coscio GC, Anelli S, Macchia E, Concetti R, Miccoli P, Arganini M, Pinchera A. Thyroid carcinoma in thyrotoxic patients treated by surgery. J Endocrinol Invest 11:107, 1988.
202. Mazzaferri, E 1990. Thyroid cancer and Graves' disease. J Clin Endocrinol Metab 70:826.
203. Sampson, R, Woolner , L, Bahn, R, Kurland, L 1974. Occult thyroid carcinoma in Olmsted County; Minnesota. Prevalence at autopsy compared with that in Hiroshima and Nagasaki; Japan. Cancer 34:2072.
204. Fukunaga, F, Yatani, R 1975. Geographic pathology of occult thyroid carcinomas. Cancer 36:1095.
205. Hazard, J, Hawk, W, Crile, G J 1959. Medullary (solid) carcinoma of the thyroid. A clinicopathologic entity. J Clin Endocrinol Metab 19:152.
206. Khurana R,. Agarwal A, Bajpai VK, Verma N, Sharma AK, Gupta RP, Madhusudan KP.Unraveling the amyloid associated with human medullary thyroid carcinoma., Endocrinology. 2004 Dec;145(12):5465-70.
207. Meier, D, Woolner, L, Beahrs, O, McConahey, W 1959. Parenchymal findings in thyroid carcinoma. Pathologic study of 256 cases. J Clin Endocrinol Metab 19:162.
208. Lindsay, S, Dailey, M 1955. Malignant lymphoma of the thyroid gland and its relation to Hashimoto's disease. A clinical and pathologic study of 8 patients. J Clin Endocrinol Metab 15:1332.
209. Rayfield, E, Nishiyama, R, Sisson, J 1971. Small cell tumors of the thyroid. A clinicopathologic study. Cancer 28:1023.
210. Black, B, Kirk, T J, Woolner, L 1960. Multicentricity of papillary adenocarcinoma of the thyroid. Influence on treatment. J Clin Endocrinol Metab 20:130.
211. Iida, F, Yonekura, M, Miyakawa, M 1969. Study of intraglandular dissemination of thyroid cancer. Cancer 24:764.
212. Reed, R, Russin, D, Krementz, ET. 1966. Latent metastases from occult sclerosing carcinoma of the thyroid. JAMA 196:233.
213. Hazard, J 1960. Small papillary carcinoma of the thyroid. Lab Invest 9:86.
214. Chung AY, Tran TB, Brumund KT, Weisman RA, Bouvet M. 2012 Metastases to the thyroid: a review of the literature from the last decade. Thyroid. 22:258-68.
215. Studer, H, Veraguth, P, Wyss, F 1961. Thyrotoxicosis due to a solitary hepatic metastasis of thyroid carcinoma. J Clin Endocrinol Metab 21:1334.
216. Hunt, W, Crispell, K, McKee, J 1960. Functioning metastatic carcinoma of the thyroid producing clinical hyperthyroidism. Am J Med 28:995.
217. Pochin, E 1960. Leukemia following radioiodine treatment of thyrotoxicosis. Br Med J 2:1545.
218. Wyse, E, Hill, C, Ibanez, M, Clark, R 1969. Other malignant neoplasms associated with carcinoma of the thyroid. Thyroid carcinoma multiplex. Cancer 24:701.
219. Shimaoka, K, Takeuchi, S, Pickren, J 1967. Carcinoma of thyroid associated with other primary malignant tumors. Cancer 20:1000.
220. Halnan, K 1966. Influence of age and sex on incidence and prognosis of thyroid cancer. Cancer 19:1534.
221. Russel, M, Gilbert, E, Jaeschke, W 1975. Prognostic features of thyroid cancer. A long term follow-up of 68 cases. Cancer 36:553.
222. Rosvoll, R, Winship, T 1965. Thyroid carcinoma and pregnancy. Surg Gynecol Obstet 121:1039.
223. Franssila, K 1975. Prognosis in thyroid carcinoma. Cancer 36:1138.
224. Woolner, L, Lemmon, M, Beahrs, O, Black, B, Keating, F J 1960. Occult papillary carcinoma of the thyroid. Study of 140 cases observed in a 30-year period. J Clin Endocrinol Metab 20:89-113.
225. Passler, C; Prager, G; Scheuba, C; Niederle, BE; Kaserer, K; Zettinig, G; Niederle, B. Follicular variant of papillary thyroid carcinoma: a long-term follow-up. Arch Surg 138:1362-1366,2003.
226. Shattuck TM, Westra WH, Ladenson PW, Arnold A. Independent clonal origins of distinct tumor foci in multifocal papillary thyroid carcinoma. N Engl J Med. 2005, 352:2406-12.
227. McConahey, W, Taylor, W, Gorman, C, Woolner, L Field. Educational Italian Retrospective study of 820 patients treated for papillary carcinoma of the thyroid at the Mayo Clinic between 1946 and 1971. In Andreoli, M; Monaco, F; Robbins, J (eds). Advances in Thyroid Neoplasia: Rome.
228. C. Ceccarelli, F. Pacini, F. Lippi, R. Elisei, M. Arganini, P. Miccoli, A. Pinchera. Thyroid cancer in children and adolescent. Surgery 104:1143, 1988.
229. McDermott, W J, Morgan, W, Hamlin, E J, Cope, O 1954. Cancer of the thyroid. J Clin Endocrinol Metab 14:1336.
230. Schlumberger, M, De Vathaire, F, Travagli, J, et al. 1987. Differentiated thyroid carcinoma in childhood. Long term follow-up of 72 patients. J Clin Endocrinol Metab 65:1088.
231. Cady, B, Sedgwick, C, Meissner, W, Bookwalter, J, Romagosa, V, Werber, J 1976. Changing clinical; pathologic; therapeutic; and survival patterns of differentiated thyroid carcinoma. Ann Surg 184:541.
232. Mazzaferri, E, Young, R 1981. Papillary thyroid carcinoma. A ten year follow-up report of the impact of therapy in 576 patients. Am J Med 70:511.
233. Harwood, J, Clark, O, Dunphy, J 1978. Significance of lymph node metastasis in differentiated thyroid cancer. Am J Surg 136:107.
234. Mazzaferri, E, Young, R, Oertel, J, Kemmerer, W, Page, C 1977. Papillary thyroid carcinoma. The impact of therapy in 576 patients. Medicine 56:171.
235. Terry, J, St John, S, Karkowski, F, al., e 1994. Tall cell papillary thyroid cancer. Incidence and prognosis. Am J Surg 168:459.
236. Burman, K, Ringel, M, Wartofsky, L 1996. Unusual types of thyroid neoplasms. Endocrinol Metab Clinics North America 25:49-68.
237. Frazell, E, Duffy, B 1954. Invasive papillary cancer of the thyroid. J Clin Endocrinol Metab 14:1362.
238. Grebe SK, Hay ID. Follicular thyroid cancer. Endocrinol Metab Clin North Am. 1995, 24:761-801.
239. Elisei R, Molinaro E, Agate L, Bottici V, Masserini L, Ceccarelli C, Lippi F, Grasso L, Basolo F, Bevilacqua G, Miccoli P, Di Coscio G, Vitti P, Pacini F, Pinchera A. Are the clinical and pathological features of differentiated thyroid carcinoma really changed over the last 35 years? Study on 4187 patients from a single Italian institution to answer this question. J Clin Endocrinol Metab. 2010, 95:1516-27.
240. Chao, TC; Lin, JD; Chen, MF. Insular carcinoma: Infrequent subtype of thyroid cancer with aggressive clinical course. World J Surg, 2004, 28:393-6.
241. Young, R, Mazzaferri, E, Rahe, A, Dorfman, S 1980. Pure follicular thyroid carcinoma. Impact of therapy in 214 patients. J Nucl Med 21:733.
242. Justin, E, Seabold, J, Robinson, R, Walker, W, Gurll, N, Hawe, D 1991. Insular carcinoma. A distinct thyroid carcinoma with associated Iodine-131 localization. J Nucl Med 32:1358-1.
243. Caplan, R, Abellera, R, Kisken, W 1994. Hurthle cell neoplasms of the thyroid gland. Reassessment of functional capacity. Thyroid 4:243.
244. Cheung, C, Ezzat, S, Ramyar, L, Freeman, J, Asa, S 2000. Molecular basis of Hurthle cell papillary thyroid carcinoma. J Clin Endocrinol Metab 85:878-882.
245. Harada, T, Ito, K, Shimaoka, K, Hosoda, Y, Yakumara, K 1977. Fatal thyroid carcinoma. Anaplastic transformation of adenocarcinoma. Cancer 39:2588.
246. Oppenheim, al., e 1983. Analplastic thyroid cancer presenting with hyperthyroidism. Amer J Med 75:702.
247. Leedman, P, Sheridan, W, Downey, W, Fox, R, Martin, F 1990. Combination chemotherapy as single modality therapy for stage IE and IIE thyroid lymphoma. Med J Australia 152:40.
248. Butler, J, Brady, L, Amendola, B 1990. Lymphoma of the thyroid. Report of five cases and review. Amer J Clin Oncol (CCT) 13:64.
249. Souhami, L, Simpson, W, Carrothers, J 1980. Malignant lymphoma of the thyroid gland. Int J Radiat Oncol Biol Phys 6:1143.
250. Kini, S, Miller, J, Hamburger, J 1981. Problems in the cytologic diagnosis of the "cold" thyroid nodule in patients with lymphocytic thyroiditis. Acta Cytol 25:506.
251. Grimley, R, Oates, G 1980. The natural history of malignant thyroid lymphomas. Br J Surg 67:475.
252. Siroto, D, Segal, R 1979. Primary lymphomas of the thyroid. JAMA 242:1743.
253. Cignarelli, M, Ambrosi, A, Marino, A, Lamacchia, O, Cincione, R, Neri, V 2002 Three cases of papillary carcinoma and three of adenoma in thyroglossal duct cysts: clinical-diagnostic comparison with benign thyroglossal duct cysts. J Endocrinol Invest 25:947-54.
254. Moline J, Eng C. Multiple endocrine neoplasia type 2: an overview. 2011. Genet Med. 2011 13:755-64.
255. Sobol, H, Narod, S N, Y, Boneu, A, et al. 1989. Screening for multiple endocrine neoplasia type 2a with DNA-polymorphism analysis. N Engl J Med 321:996-1001.
256. Manning, P, Molnar, G, Black, M, Priestley, J, Woolner, L 1963. Pheochromocytoma; hyperparathyroidism; and thyroid carcinoma occurring coincidentally. N Engl J Med 268:68.
257. Gorlin, R, Sedano, H, Vickers, R, Cervenka, J 1968. Multiple mucosal neuromas; pheochromocytoma; and medullary carcinoma of the thyroid -- a syndrome. Cancer 22:293
258. Gagel, R, Robinson, M, Donovan, D, Alford, B 1993. Medullary thyroid carcinoma. Recent progress. J Clin Endocrinol Metab 76:.809-814.
259. Carney, J, Go, V, Sizemore, G, Hayles, A 1976. Alimentary-tract ganglioneuromatosis. N Engl J Med 295:1287.
260. Noel, M, Delehaye, M-C, Segond, N, et al. 1991. Study of calcitonin and thyroglobulin gene expression in human mixed follicular and medullary thyroid carcinoma. Thyroid 1:249.
261. Wolfe, H, Melvin, K, Cervi-Skinner, S, et al. 1973. C-cell hyperplasia preceding medullary thyroid carcinoma. N Engl J Med 289:437.
262. Pacini, F, Basolo, F, Elise, R, Fugazzola, L, Cola, A, Pinchera, A 1991. Medullary thyroid cancer. An immunohistochemical and humoral study using six separate antigens. Amer J Clin Pathol. 95:300.
263. Kanamoto N, Akamizu T, Hosoda H, Hataya Y, Ariyasu H, Takaya K, Hosoda K, Saijo M, Moriyama K, Shimatsu A, Kojima M, Kangawa K, and Nakao K. Substantial production of Ghrelin by a human medullary thyroid carcinoma cell line. J Clin Endocrinol Metab 86:4984-4990, 2001.
264. Cohen, S, Graham-Smith, D, MacIntyre, I, Walker, J 1973. Alcohol- stimulated calcitonin release in medullary carcinoma of the thyroid. Lancet 2:1172.
265. Melvin, K, Tashjian, A 1968. The syndrome of excessive thyrocalcitonin produced by medullary carcinoma of the thyroid. Proc Natl Acad Sci USA 59:1216.
266. Elisei R, Cosci B, Romei C, Bottici V, Renzini G, Molinaro E, Agate L, Vivaldi A, Faviana P, Basolo F, Miccoli P, Berti P, Pacini F, Pinchera A. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008, 93:682-7.
267. Melvin, K, Miller, H, Tashjian, A 1971. Early diagnosis of medullary carcinoma of the thyroid gland by means of calcitonin assay. N Engl J Med 285:1115.
268. Richard T. Kloos, Charis Eng, Douglas B. Evans, Gary L. Francis, Robert F. Gagel, Hossein Gharib, Jeffrey F. Moley, Furio Pacini, Matthew D. Ringel, Martin Schlumberger, and Samuel A. Wells Jr. 2009. Medullary Thyroid Cancer: Management Guidelines of the American Thyroid Association. Thyroid 19:565.
269. Barbot, N, Calmettes, C, Schuffenecker, I, et al. 1994. Pentagastrin stimulation test and early diagnosis of medullary thyroid carcinoma using an immunoradiometric assay of calcitonin. Comparison with genetic screening in hereditary medullary thyroid carcinoma. J Clin Endocrinol Metab 78:114-120.
270. Calmettes, C, Moukhtar, M, Milhaud, G 1977. Correlation between calcitonin and carcinoembryonic antigen levels in medullary carcinoma of the thyroid. Biomedicine 27:52.
271. Hoefnagel, C, Delprat, C, Zanin, D, Van Der Schoot, J 1988. New radionuclide tracers for the diagnosis and therapy of medullary thyroid carcinoma. Clin Nucl Med 13:159.
272. Pacini, F, Ceccherini, I, Martino, E, et al. Abstract No. 182. Screening for ret gene mutations in multiple endocrine neoplasia (MEN) type 2 and in sporadic medullary thyroid carcinoma (MTC). clinical applications. Sixty-eighth annual meeting of the American Thyroid Association Chicago; IL: September 28-October 1; 1994.
273. Lips, C, Landsvater, R, Hoppener, J, et al. 1994. Clinical screening as compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N Engl J Med 331:828-835.
274. Black, H, Capen, C, Young, D 1973. Ultimobranchial thyroid neoplasms in bulls. Cancer 32:865.
275. Graze, K, Spiler, I, Tashijan, A J, et al. 1978. Natural history of familial medullary thyroid carcinoma. N Engl J Med 299:980.
276. Machens, A; Niccoli-Sire, P; Hoegel, J; Frank-Raue, K; van Vroonhoven, TJ; Roeher, H-D; Wahl, RA; Lamesch, P; Raue, F; Conte-Devolx, B; Dralle, H. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med 349 1517-1525 2003.
277. Scollo C, Baudin E, Travagli J-P, Caillou B, Bellon N, Leboulleux S, Schlumberger M. Rationale for central and bilateral lymph node dissection in sporadic and hereditary medullary thyroid cancer. J Clin Endocrinol Metab 88:2070-2075, 2003.
278. Deftos, L, Stein, M 1980. Radioiodine as an adjunct to the surgical treatment of medullary thyroid carcinoma. J Clin Endocrinol Metab 50:967.
279. Hellman, D, Kartchner, M, Van Antwerp, J, Salmon, S, Patton, D, O'Mara, R 1979. Radioiodine in the treatment of medullary carcinoma of the thyroid. J Clin Endocrinol Metab 48:451.
280. Kim, S, Morimoto, S, Kawa, i Y, Koh, E, Onishi, T, Ogihara, T 1989. Circulating levels of calcitonin gene-related peptide in patients with medullary thyroid carcinoma. J Clin Chem Clin Biochem 27:423
281. Moley, J, Wells, S, Dilley, W, Tisell, L 1993. Reoperation for recurrent or persistent medullary thyroid cancer. Surgery 114:1090-1095.
282. Frank-Raue, K, Raue, F, Buhr, H, Baldauf, G, Lorenz, D, Ziegler, R 1992. Localization of occult persisting medullary thyroid carcinoma before microsurgical reoperation. High sensitivity of selective venous catheterization. Thyroid 2:113.
283. Simpson, W 1975. Radiotherapy in thyroid cancer. Can Med Assoc J 113:115.
284. Brierley, J, Tsang, R, Simpson, W, Gospodarowicz, M, Sutcliffe, S, Panzarella, T 1996. Medullary thyroid cancer. Analyses of survival and prognostic factors and the role of radiation therapy in local control. Thyroid 6:305-310.
285. Fersht, N, Vini, L, A'Hern, R, Harmer, C 2001 The role of radiotherapy in the management of elevated calcitonin after surgery for medullary thyroid cancer. Thyroid 11:1161-8.
286. Samaan, N, Schultz, P, Hickey, R Medullary thyroid carcinoma. Prognosis of familial versus nonfamilial disease and the role of radiotherapy.
287. DeGroot, L 1970. Lack of iodide trapping in "cold" thyroid nodules. Acta Endocrinol Panam 1:27.
288. Kim, B W, Daniels, G H, Harrison, B J, et al. 2003 Overexpression of type 2 iodothyronine deiodinase in follicular carcinoma as a cause of low circulating free thyroxine levels. J Clin Endocrinol Metab 88:594-8.
289. Huang, S A, Fish, S A, Dorfman, D M, et al. 2002 A 21-year-old woman with consumptive hypothyroidism due to a vascular tumor expressing type 3 iodothyronine deiodinase. J Clin Endocrinol Metab 87:4457-61
290. Abe, Y, Ichikawa, Y, Muraki, T, Ito, K, Momma, M 1981. Thyrotropin (TSH) receptor and adenylate cyclase activity in human thyroid tumors. Absence of high affinity receptor and loss of TSH responsiveness in undifferentiated thyroid carcinoma. J Clin Endocrinol Metab 52:23.
291. Carayon, P, Thomas-Morvan, C, Castanas, E, Tubiana, M 1980. Human thyroid cancer. Membrane thyrotropin binding and adenylate cyclase activity. J Clin Endocrinol Metab 51:915.
292. Burke, G, Szabo, M 1972. Dissociation of in vivo and in vitro "autonomy" in hyperfunctioning thyroid nodules. J Clin Endocrinol Metab 35:199.
293. Van Sande, J, Mockel, J, Boeynaems, J, Dor, P, Andry, G, Dumont, J 1980. Regulation of cyclic nucleotide and prostaglandin formation in normal human thyroid tissue and in autonomous nodules. J Clin Endocrinol Metab 50:776.
294. Franklin, W, Mariotti, S, Kaplan, D, DeGroot, L 1982. Immunofluorescence localization of thyroglobulin in metastatic thyroid cancer. Cancer 50:939.
295. Goudie, R, McCallum, H 1963. Loss of tissue-specific autoantigens in thyroid tumors. Lancet 1:348.
296. Pontius, K, Hawk, W 1980. Loss of microsomal antigen in follicular and papillary carcinoma of the thyroid. An immunofluorescence and electron-microscope study. Am J Pathol 74:620.
297. Aoki, N, DeGroot, L 1979. Lymphocyte blastogenic response to human thyroglobulin in Graves' disease; Hashimoto;s thyroiditis; and metastatic thyroid cancer. Clin Exp Immunol 38:523.
298. Amino, N, Pysher, T, Cohen, E, DeGroot, L 1975. Immunologic aspects of human thyroid cancer. Cancer 36:963.
299. Mariotti, S, DeGroot, L, Scarborough, D, Medof, M 1979. Study of circulating immune complexes in thyroid diseases. Comparison of Raji cell radioimmunoassay and specific thyroglobulin-antithyroglobulin radioassay. J Clin Endocrinol Metab 49:679.
300. Pacini, F, Mariotti, S, Formica, N, et al. 1988 Thyroid autoantibodies in thyroid cancer. Incidence and relationship with tumour outcome. Acta Endocrinol (Copenh) 119:373.
301. Rubello, D, Casara, D, Girelli, M, Piccolo, M, Busnardo, B 1992. Clinical meaning of circulating antithyroglobulin antibodies in differentiated thyroid cancer. a prospective study. J Nucl Med 33:1478
302. Chiovato, L; Latrofa, F; Braverman, LE; Pacini, F; Capezzone, M; Masserini, L; Grasso, L; Pinchera, A. Disappearance of humoral thyroid autoimmunity after complete removal of thyroid antigens. Ann Intern Med 139:346-351, 2003.
303. Rocklin, E, Gagel, R, Feldman, Z, Tashijan, A J 1977. Cellular immune responses in familial medullary thyroid carcinoma. N Engl J Med 296:835.
304. Wu, P-C, Leslie, P, McLaren, K, Toft, A 1989. Diffuse sclerosing papillary carcinoma of thyroid. A wolf in sheep's clothing. Clin Endocrinol 31:535.
305. Van Herle, A, Uller, R 1975. Elevated serum thyroglobulin. A marker of metastases in differentiated thyroid carcinomas. J Clin Invest 56:272.
306. Cignarelli, M, Triggiani, V, Ciampolillo, A, et al. 2001. High frequency of incidental diagnosis of extrathyroidal neoplastic diseases at the fine-needle aspiration biopsy of laterocervical lymph nodes in patients with thyroid nodules. Thyroid 11:65.
307. Meighan, J, Dworkin, H 1969. Failure to detect 131I positive thyroid metastases with 99mTc. J Nucl Med 11:173.
308. Wang, W, Larson, S, Fazzari, M, et al. 2000. Prognostic value of [18F]-Fluorodeoxy-glucose positron emission tomographic scanning in patients with thyroid cancer. J Clin Endocrinol Metab 85:1107-1113.
309. Van Tol KM, Jager PL, Piers A, Pruim J, de Vries EGE, Dullaart RPF, Links TP. Better yield of 18-Fluorodeoxyglucose-positron emission tomography in patients with metastatic differentiated thyroid carcinoma during thyrotropin stimulation. Thyroid 12:381, 2002. .
310. Chin, BB; Patel, P; Cohade, C; Ewertz, M; Wahl, R; Ladenson, P. Recombinant human thyrotropin stimulation of fluoro-D-glucose positron emission tomography uptake in well-differentiated thyroid carcinoma. J Clin Endocrinol Metab 89 91-95 2004.
311. Samaan, N, Schultz, P, Hickey, R, et al. 1992. The results of various modalities of treatment of well differentiated thyroid carcinoma. A retrospective review of 1599 patients. J Clin Endocrinol Metab 75:714-720
312. Hay, I, Grant, C, Bergstralh, E, Thompson, G, van Heerden, J, Goellner, J 1998. Unilateral total lobectomy. Is it sufficient surgical treatment for patients with AMES low-risk papillary thyroid carcinoma? Surgery 124:958-966.
313. Rustad, W, Lindsay, S, Dailey, M 1963. Comparison of the incidence of complications following total and subtotal thyroidectomy for thyroid carcinoma. Surg Gynecol Obstet 116:109.
314. Thompson, N, Harness, J; 1970. Complications of total thyroidectomy for carcinoma. Surg Gynecol Obstet 131:861.
315. Tollefson, H, DeCosse, J 1964. Papillary carcinoma of the thyroid. The case for radical neck dissection. Am J Surg 108:547.
316. DeGroot LJ, Kaplan EL, McCormick M, Straus FH. Natural history, treatment, and course of papillary thyroid carcinoma. J Clin Endocrinol Metab. 1990, 71:414-24.
317. Tennvall J, Biorklund A, Moller T et al. Is the EORTC prognostic index of thyroid cancer valid in differentiated thyroid Carcinoma? Cancer 57:1405, 1986.
318. Pasieka JL, Zedenius J, Azuer G et al; Addition of nuclear content to the AMES risk-group classification for papillary thyroid cancer: Surgery 112:1154, 1992.
319. Hay ID, Bergstralh EJ, Goellner JR, et al. Predicting outcome in papillary thyroid carcinoma. Surgery 114: 1050, 1993.
320. Tuttle RM, Tala H, Shah J, Leboeuf R, Ghossein R, Gonen M, Brokhin M, Omry G, Fagin JA, Shaha A.2010 Estimating risk of recurrence in differentiated thyroid cancer after total thyroidectomy and radioactive iodine remnant ablation: using response to therapy variables to modify the initial risk estimates predicted by the new American Thyroid Association staging system. Thyroid. 20:1341-9
321. Castagna MG, Maino F, Cipri C, Belardini V, Theodoropoulou A, Cevenini G, Pacini F. 2011. Delayed risk stratification, to include the response to initial treatment (surgery and radioiodine ablation), has better outcome predictivity in differentiated thyroid cancer patients. Eur J Endocrinol. 165:441-6.
322. Klapp, C, Rosvoli, R, Winship, T 1967. Is destructive surgery ever necessary for treatment of thyroid cancer in children? Ann Surg 165:745
323. Liechty, R, Safaie-Shirazi, S, Soper, R 1972. Carcinoma of the thyroid in children. Surg Gynecol Obstet 134:595.
324. Pacini, F, Gasperi, M, Fugazzola, L, et al. 1994. Testicular function in patients with differentiated thyroid carcinoma treated with radioiodine. J Nucl Med 35:1418.
325. McClellan, D, Francis, G 1996. Thyroid cancer in children; pregnant women; and patients with Graves' disease. Endocrinol Metab Clinics North America 25:27-48.
326. Casara, D, Rubello, D, Saladini, G, et al. 1993. Pregnancy after therapeutic doses of iodine-131 in differentiated thyroid cancer. potential risks and recommendations. Eur J Nucl Med 20:192.
327. Schlumberger, M, De Vathaire, F, Ceccarelli, C, et al. 1996. Exposure to radioactive iodine-131 for scintigraphy or therapy does not preclude pregnancy in thyroid cancer patients. J Nucl Med 37:606.
328. Cady, B, Sedgwick, C, Meissner, W, Bookwalter, J, Romagosa, V, Werber, J 1976. Changing clinical; pathologic; therapeutic; and survival patterns of differentiated thyroid carcinoma. Ann Surg 184:541.
329. DeGroot, L, Reilly, M 1982. Comparison of 30- and 50-mCi doses of iodine-131 for thyroid ablation. Ann Intern Med 96:51.
330. Roos, D e a 1999. Review of trials assessing low dose radioactive iodine ablation for thyroid remnants in patients with thyroid cancer. International J Rad Oncol Biol Physiol 44:493-495.
331. Tala Jury HP, Castagna MG, Fioravanti C, Cipri C, Brianzoni E, Pacini F. 2010. Lack of association between urinary iodine excretion and successful thyroid ablation in thyroid cancer patients. J Clin Endocrinol Metab.95:230-7.
332. Karam M, Ianoukakis A, Feustel PJ, Cheema A, Postal ES, Cooper JA. Influence of diagnostic and therapeutic doses on thyroid remnant ablation rates. Nucl Med Commun 24:489-95, 2003.
333. Wartofsky, L, Sherman, S, Gopal, J, Schlumberger, M, Hay, I 1998. The use of radioactive iodine in patients with papillary and follicular thyroid cancer. J Clin Endocrinol Metab 83:4195-4203.
334. Carcangiu, M, Bianchi, S, Savino, D, Voynick, I, Rosai, J 1991. Follicular Hurthle cell tumors of the thyroid gland. Cancer 68:1944-1953.
335. Pacini, F, Molinaro, E, Castagna, M G, et al. 2002 Ablation of thyroid residues with 30 mCi (131)I: a comparison in thyroid cancer patients prepared with recombinant human TSH or thyroid hormone withdrawal. J Clin Endocrinol Metab 87:4063-8.
336. Robbins, R J, Tuttle, R M, Sonenberg, M, et al. 2001 Radioiodine ablation of thyroid remnants after preparation with recombinant human thyrotropin. Thyroid 11:865-9.
337. Pacini F, Ladenson PW, Schlumberger M, Driedger A, Luster M, Kloos RT, Sherman S, Haugen B, Corone C, Molinaro E, Elisei R, Ceccarelli C, Pinchera A, Wahl RL, Leboulleux S, Ricard M, Yoo J, Busaidy NL, Delpassand E, Hanscheid H, Felbinger R, Lassmann M, Reiners C. Radioiodine ablation of thyroid remnants after preparation with recombinant human thyrotropin in differentiated thyroid carcinoma: results of an international, randomized, controlled study. J Clin Endocrinol Metab. 2006, 91:926-32.
338. Guimaraes V, DeGroot LJ. Moderate hypothyroidism in preparation for whole body 131I scintiscans and thyroglobulin testing. Thyroid. 1996, 6:69-73
339. Ramirez, L, Braverman, L, White, B, Emerson, C 1997. Recombinant human thyrotropin is a potent stimulator of thyroid function in normal subjects. J Clin Endocrinol Metab 82:2836-2839.
340. Meier, C, Braverman, L, Ebner, S, et al. 1994. Diagnostic use of recombinant human thyrotropin in patients with thyroid carcinoma (phase I/II study). J Clin Endocrinol Metab; 78:188-96.
341. Ladenson, P, Braverman, L, Mazzaferri, E, al., e 1997. Comparison of administration of recombinant human thyrotropin with withdrawal of thyroid hormone for radioactive iodine scanning in patients with thyroid carcinoma. N Engl J Med 337:888-95.
342. Haugen, B, Pacini, F, Reiners, C, et al. 1999. A comparison of recombinant human thyrotropin and thyroid hormone withdrawal for the detection of thyroid remnant or cancer. J Clin Endocrinol Metab 84:3877-3885.
343. Driedger, AA; Kotowycz, N. Two cases of thyroid carcinoma that were not stimulated by recombinant human thyrotropin. J Clin Endocrinol Metab 89 585-590 2004.
344. Sorvillo, F; Mazziotti,G; Carbone, A; Piscopo, M; Rotondi, M; Cioffi, M; Musto, P; Biondi, B; Iorio, S; Amato, G; Carella, C. Recombinant human thyrotropin reduces serum vascular endothelial growth factor levels in patients monitored for thyroid carcinoma even in the absence of thyroid tissue. J Clin Endocrinol Metab 88 4818-4822 2003.
345. Luster M, Lippi F, Jarzab B, Perros P, Lassmann M, Reiners C, Pacini F. rhTSH-aided radioiodine ablation and treatment of differentiated thyroid carcinoma: a comprehensive review.Endocr Relat Cancer. 2005 Mar;12(1):49-64.
346. Bachelot, A, Cailleux, A F, Klain, M, et al. 2002 Relationship between tumor burden and serum thyroglobulin level in patients with papillary and follicular thyroid carcinoma. Thyroid 12:707-11.
347. Pacini, F, Capezzone, M, Elisei, R, Ceccarelli, C, Taddei, D, Pinchera, A 2002 Diagnostic 131-iodine whole-body scan may be avoided in thyroid cancer patients who have undetectable stimulated serum Tg levels after initial treatment. J Clin Endocrinol Metab 87:1499-501.
348. Pacini F, Molinaro E, Lippi F, Castagna MG, Agate L, Ceccarelli C, Taddei D, Elisei R, Capezzone M, and Pinchera A. Prediction of disease status by recombinant human TSH-stimulated serum Tg in the postsurgical follow-up of differentiated thyroid carcinoma. J Clin Endocrinol Metab 86:5686-5690, 2001.
349. Mazzaferri, E, Kloos, R 2002. Is diagnostic Iodine-131 scanning with recombinant human TSH useful in the follow-up of differentiated thyroid cancer after thyroid ablation? J Clin Endocrinol Metab 87:1490-1498.
350. Wartofsky, L 2002. Using baseline and recombinant human TSH-stimulated tg measurements to manage thyroid cancer without diagnostic 131I scanning. J Clin Endocrinol Metab 87:1486-1489.
351. Wartofsky, L 2002 Management of low-risk well-differentiated thyroid cancer based only on thyroglobulin measurement after recombinant human thyrotropin. Thyroid 12:583-90.
352. Wartofsky L; rhTSH-Stimulated Thyroglobulin Study Group. Management of low-risk well-differentiated thyroid cancer based only on thyroglobulin measurement after recombinant human thyrotropin. Thyroid. 2002, 12:583-90
353. Pacini F, Schlumberger M, Dralle H, Elisei R, Smit JWA, Wiersinga W and the European Thyroid Cancer Taskforce. European consensus for the management of patients with differentiated thyroid carcinoma of the follicular epithelium. Eur J Endocrinol, 154:1-18, 2006.
354. Park, H-M, Perkins, O, Edmondson, J, Schnute, R, Manatunga, A 1994. Influence of diagnostic radioiodines on the uptake of ablative dose of Iodine-131. Thyroid 4:49.
355. Lassmann,M Luster M, Hanscheid H, Reiners C. Impact of 1313-I diagnostic Activities on the Biokinetics of Thyroid Remnants. J Nucl Med 45: 619-625, 2004.
356. Morris, L F, Waxman, A D, Braunstein, G D 2001 The nonimpact of thyroid stunning: remnant ablation rates in 131I-scanned and nonscanned individuals. J Clin Endocrinol Metab 86:3507-11.
357. Cailleux, A, Baudin, E, Travagli, J, Ricard, M, Schlumberger, M 2000. Is diagnostic Iodine-131 scanning useful after total thyroid ablation for differentiated thyroid cancer? J Clin Endocrinol Metab 85:175-178.
358. Pacini F, Capezzone M, Elisei R, Ceccarelli C, Taddei D, Pinchera A. Diagnostic 131-Iodine whole-body scan may be avoided in thyroid cancer patients who have undetectable stimulated serum Tg levels after initial treatment. J Clin Endocrinol Metab 87:1499-1501, 2002.
359. Haq MS, MacCready RV, Harmer CL. Treatment of advanced differentiated thyroid carcinoma with high activity radioiodine therapy. Nucl Med Commun 25:799-805, 2004.
360. Dorn R, Kopp J, Vogt H, Heidenreich P, Carroll RG, Gulec SA.Dosimetry-guided radioactive iodine treatment in patients with metastatic differentiated thyroid cancer: largest safe dose using a risk-adapted approach. J Nucl Med 44:4516, 2003.
361. Van Nostrand, D, Atkins, F, Yeganeh, F, Acio, E, Bursaw, R, Wartofsky, L 2002. Dosimetrically determined doses of radioiodine for the treatment of metastatic thyroid carcinoma. Thyroid 12:121-134.
362. O'Connell, M, Flower, M, Hinton, P, Harmer, C, McCready, V 1993. Radiation dose assessment in radioiodine therapy. Dose-response relationships in differentiated thyroid carcinoma using quantitative scanning and PET. Radiotherapy-Oncology 28:16-26.
363. Maxon, H, Thomas, S, Hertzberg, V, et al. 1983. Relation between effective radiation dose and outcome of radioiodine therapy for thyroid cancer. N Engl J Med 309:937.
364. Koong, S-S, Reynolds, J, Movius, E, et al. 1999. Lithium as a potential adjuvant to 131I therapy of metastatic; well differentiated thyroid carcinoma. J Clin Endocrinol Metab 84:912-916.
365. Fatourechi, V, Hay, I, Mullan, B, et al. 2000. Are posttherapy radioiodine scans informative and do they influence subsequent therapy of patients with differentiated thyroid cancer? Thyroid 10:573.
366. Pelikan, D, Lion, H, Hermans, J, Goslings, B 1997. The role of radioactive iodine in the treatment of advanced differentiated thyroid carcinoma. Clin Endocrinol 47:713-720.
367. Menzel, C, Grunwald, F, Schomburg, A, et al. 1996. "High-dose" radioiodine therapy in advanced differentiated thyroid carcinoma. J Nucl Med 37:1496-1503.
368. Taylor, T, Specker, B, Robbins, J, et al. 1998. Outcome after treatment of high-risk papillary and non-Hurthle-cell follicular thyroid carcinoma. Ann Intern Med 129:622-627.
369. Pittas, A, Adler, M, Fazzari, M, et al. 2000. Bone metastases from thyroid carcinoma. clinical characteristics and prognostic variables in one hundred forty-six patients. Thyroid 10:261-268.
370. Van Tol, K, Hew, J, Jager, P, Vermey, A, Dullaart, R, Links, T 2000. .Embolization in combination with radioiodine therapy for bone metastases from differentiated thyroid carcinoma. Clin Endocrinol 52:653-659.
371. Hamburger, J, Desai, P Mannitol augmentation of I131 uptake in the treatment of thyroid carcinoma. Metabolism 15 1055: 1966.
372. Hamburger, J 1969. Diuretic augmentation of 131I uptake in inoperable thyroid cancer. N Engl J Med 280:1091.
373. Pacini, F, Lippi, F, Formica, N, et al. 1987. Therapeutic doses of iodine-131 reveal undiagnosed metastases in thyroid cancer patients with detectable serum thyroglobulin levels. J Nucl Med 28:1888.
374. Koh J-M, Kim ES, Ryu JS, Hong SJ, Kim WB, Shong YK. Effects of therapeutic doses of 131I in thyroid papillary carcinoma patients with elevated thyroglobulin level and negative 131I whole-body scan: comparative study. Clin Endocrinol 58:421-427, 2003.
375. Schlumberger, M, Arcangioli, O, Piekarski, J, Tubiana, M, Parmentier, C 1988. Detection and treatment of lung metastases of differentiated thyroid carcinoma in patients with normal chest X-rays. J Nucl Med 29:1790-1794.
376. Pineda, J, Lee, T, Ain, K, Reynolds, J, Robbins, J 1995. Iodine-131 therapy for thyroid cancer patients with elevated thyroglobulin and negative diagnostic scan. J Clin Endocrinol Metab 80:1488.
377. McDougall, I 1997 131I treatment of 131I negative whole body scan; and positive thyroglobulin in differentiated thyroid carcinoma. what is being treated? Thyroid 7:669.
378. Fatourechi, V, Hay, I D, Javedan, H, Wiseman, G A, Mullan, B P, Gorman, C A 2002 Lack of impact of radioiodine therapy in tg-positive, diagnostic whole-body scan-negative patients with follicular cell-derived thyroid cancer. J Clin Endocrinol Metab 87:1521-6.
379. Grigsby, P, Siegel, B, Baker, S, Eichling, J 2000. Radiation exposure from outpatient radioactive iodine (131I) therapy for thyroid carcinoma. J Amer Med Assn 283:2272-2274.
380. American Thyroid Association Taskforce On Radioiodine Safety, Sisson JC, Freitas J, McDougall IR, Dauer LT, Hurley JR, Brierley JD, Edinboro CH, Rosenthal D, Thomas MJ, Wexler JA, Asamoah E, Avram AM, Milas M, Greenlee C. Radiation safety in the treatment of patients with thyroid diseases by radioiodine 131I : practice recommendations of the American Thyroid Association. Thyroid. 2011, 21:335-46. Erratum in: Thyroid. 2011, 21:689.
381. Seidlin, S, Yalow, R, Siegel, E 1952. Blood radioiodine concentration and blood radiation dosage during I131 therapy for metastatic thyroid carcinoma. J Clin Endocrinol Metab 12:1197.
382. Mandel SJ, Mandel L. Radioactive iodine and the salivary glands. Thyroid 13:265-271, 2003.
383. Raymond, J, Izembart, M, Marliac, V, et al. 1989. Temporary ovarian failure in thyroid cancer patients after thyroid remnant ablation with radioactive iodine. J Clin Endocrinol Metab 69:186.
384. Ceccarelli, C, Battisti, P, Gasperi, M, et al. 1999. Radiation dose to the testes after 131I therapy for ablation of postsurgical thyroid remnants in patients with differentiated thyroid cancer. J Nucl Med 40:1716.
385. Wiseman, J, Hales, I, Joasoo, A 1982. Two cases of lymphoma of the parotid gland following ablative radioiodine therapy for thyroid carcinoma. Clin Endocrinol 17:85.
386. Rall, J, Alpers, J, Lewallen, C, Sonenberg, M, Berman, M, Rawson, R 1957. Radiation pneumonitis and fibrosis. A complication of I131 treatment of pulmonary metastases from cancer of the thyroid. J Clin Endocrinol Metab 17:1263.
387. Trunnell, J, Marinelli, L, Duffy, B J, Hill, R, Peacock, W, Rawson, R 1949. The treatment of metastatic thyroid cancer with radioactive iodine. Credits and debits. J Clin Endocrinol Metab 19:1138.
388. Lin, J, Wang, H, Weng, H, Kao, P 1998. Outcome of pregnancy after radioactive iodine treatment for well differentiated thyroid carcinomas. J Endocrinol Invest 21:662-667.
389. Varma, V, Beierwaltes, W, Nofal, M, Nishiyama, R, Copp, J 1437 Treatment of thyroid cancer. Death rates after surgery and after surgery followed by sodium iodide. I131 JAMA: 214.
390. Leeper, R 1973. The effect of 131I therapy on survival of patients with metastatic papillary or follicular thyroid carcinoma. J Clin Endocrinol Metab 36:1143
391. Marcocci, C, Pacini, F, Elisei, R, et al. 1989. Clinical and biologic behavior of bone metastases from differentiated thyroid carcinoma. Surgery 106:960.
392. Saenger, E, Barrett, C, Passino, J, Seltzer, R, Dooley, W 1964. Experiences with I131 in the management of carcinoma of the thyroid. Radiology 83:892.
393. Harness, J, Thompson, N, Sisson, J, Beierwaltes, W 1974. Differentiated thyroid carcinomas. Treatment of distant metastases. Arch Surg 108:410.
394. Solomon, B, Wartofsky, L, Burman, K 1996. Current trends in the management of well differentiated papillary thyroid carcinoma. J Clin Endocrinol Metab 81:333-339.
395. Shands, W, Gatling, R 1970. Cancer of the thyroid. Review of 109 cases. Ann Surg 171:735.
396. Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP, Caillou B, Ricard M, Lumbroso JD, De Vathaire F, Schlumberger M. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006, 91:2892-9.
397. Schlumberger, M 1998. Papillary and follicular thyroid carcinoma. N Engl J Med 338:.297-306.
398. Sherman, S, Tielens, E, Sostre, S, Wharam, M J, Ladenson, P 1994. Clinical utility of post-treatment radioiodine scans in the management of patients with thyroid cancer. J Clin Endocrinol Metab 78:629.
399. Capezzone, M, Sculli, M, Agate, L, Ceccarelli, C, Pacini, F 2000. Diagnostic 131-I whole body scan after total thyroidectomy and thyroid ablation is useless in thyroid cancer patients with undetectable serum thyroglobulin off l-thyroxine therapy. J Endocrinol Invest (Suppl) 23:3.
400. 515. Charles, M, Dodson, L, Waldeck, N, et al. 1980. Serum thyroglobulin levels predict total body iodine scan findings in patients with treated well-differentiated thyroid carcinoma. Am J Med 69:401.
401. Pacini, F, Pinchera, A, Giani, C, Grasso, L, Baschieri, L 1980. Serum thyroglobulin concentrations and 131-I whole body scans in the diagnosis of metastases from differentiated thyroid carcinoma (after thyroidectomy). Clin Endocrinol 13:107.
402. Barsano, C, Skosey, C, DeGroot, L, Refetoff, S 1982. Serum thyroglobulin in the management of patients with thyroid cancer. Arch Intern Med 142:763.
403. Pacini, F, Pinchera, A, Giani, C, Grasso, L, Doveri, F, Baschieri, L 1980. Serum thyroglobulin in thyroid carcinoma and other thyroid disorders. J Endocrinol Invest 3:283.
404. Johansen, K, Woodhouse, N 1992. Comparison of thyroglobulin and radioiodine scintigraphy during follow-up of patients with differentiated thyroid carcinoma. Eur J Med 1:403-406.
405. Fugazzola, L, Mihalich, A, Persani, L, et al. 2002 Highly sensitive serum thyroglobulin and circulating thyroglobulin mRNA evaluations in the management of patients with differentiated thyroid cancer in apparent remission. J Clin Endocrinol Metab 87:3201-8.
406. Elisei R, Vivaldi A, Agate L, Molinaro E, Nencetti C, Grasso L, Pinchera A, Pacini F. Low specificity of blood thyroglobulin messenger ribonucleic acid assay prevents its use in the follow-up of differentiated thyroid cancer patients. J Clin Endocrinol Metab 89:33-9, 2004.
407. Pujol, P, Daures, J-P, Nsakala, N, Baldet, L, Bringer, J, Jaffiol, C 1996. Degree of thyrotropin suppression as a prognostic determinant in differentiated thyroid cancer. J Clin Endocrinol Metab 81:4318-4323.
408. Cooper, D, Specker, B, Ho, M, et al. Thyrotropin suppression and disease progression in patients with differentiated thyroid cancer: results from the National Thyroid Cancer Treatment Cooperative Registry. 1998 Thyroid 8: 737.
409. Marcocci, C, Golia, F, Bruno-Bossio, G, Vignali, E, Pinchera, A 1994. Carefully monitored lecothyroxine suppressive therapy is not associated with bone loss in premenopausal women. . J Clin Endocrinol Metab 78:818-23.
410. Kim, J, Leeper, R 1983. Treatment of anaplastic and spindle cell carcinoma of the thyroid gland with combination adriamycin and radiation therapy. Cancer 52:954.
411. De Crevoisier R, Baudin E, Bachelot A, Leboulleux S, Travagli JP, Caillou B,Schlumberger M. Combined treatment of anaplastic thyroid carcinoma with surgery, chemotherapy,and hyperfractionated accelerated external radiotherapy. Int J Radiat Oncol Biol Phys. 2004 60:1137-43.
412. Ain, K 1998. Anaplastic thyroid carcinoma. Behavior; biology; and therapeutic approaches. Thyroid 8:715.
413. Leedman, P, Sheridan, W, Downey, W, Fox, R, Martin, F 1990. Combination chemotherapy as single modality therapy for stage IE and IIE thyroid lymphoma. Med J Australia 152:40.
414. Chak, L, Hoppe, R, Burke, J, Kaplan, H 1981. Non- Hodgkin's lymphoma presenting as thyroid enlargement. Cancer 48:2712.
415. Matsuzuka, F, Miyauchi, A, Katayama, S, et al. 1993. Clinical aspects of primary thyroid lymphoma. Diagnosis and treatment based on our experience of 119 cases. Thyroid 3:93-99.
416. Tubiana, M 1981. External radiotherapy and radioiodine in the treatment of thyroid cancer. World J Surg 5:75.
417. Riccabona, G 1979. Radiotherapy and nuclear medicine in malignant tumors of the thyroid gland. Therapiewoche 29:3448.
418. Tubiana, M, Lacour, J, Monnier, J, et al. 1975. External radiotherapy and radioiodine in the treatment of 359 thyroid cancers. Br J Radiol 48:894.
419. Brierley, J, Tsang, R 1996. External radiation therapy in the treatment of thyroid malignancy. Endocrinol Metab Clinics North America 25:141-157.
420. Lin, J-D, Tsang, N-M, Huang, M-J, Weng, H-F 1997. Results of external beam radiotherapy in patients with well differentiated thyroid carcinoma. Jpn J Clin Oncol 27:244-247.
421. Farahati, J, Reiners, C, Stuschke, M, et al. 1996. Differentiated thyroid cancer. Impact of adjuvant external radiotherapy in patients with perithyroidal tumor infiltration (Stage pT4). Cancer 77:172-179.
422. Tsang, R, Brierley, J, Simpson, W, Panzarella, T, Gospodarowicz, M, Sutcliffe, S 1998. The effects of surgery; radioiodine; and external radiation therapy on the clinical outcome of patients with differentiated thyroid carcinoma. Cancer 82:375-387.
423. Foote RL, Brown PD, Garces YI, McIver B, Kasperbauer JL. Int J Radiat Oncol Biol Phys. 2003 Jul 15;56(4):1067-72. Is there a role for radiation therapy in the management of Hurthle cell carcinoma?
424. Harada, T, Nishikawa, Y, Suzuki, T, Ito, K, Baba, S 1971. Bleomycin treatment for cancer of the thyroid. Am J Surg 122:53.
425. Gottlieb, J, Hill, C, Ibanez, M, Clark, R 1972. Chemotherapy of thyroid cancer. An evaluation of experience with 37 patients. Cancer 30:848.
426. Jereb, B, Stjernsward, J, Lowhagen, T 1975. Anaplastic giant-cell carcinoma of the thyroid. Cancer 35:1293.
427. Shimaoka, K, Reyes, J American Elsevier Chemotherapy of thyroid carcinoma. in Robbins, J; Braverman, L (eds). Thyroid Research: New York.
428. Riccabona, G, Zechmann, W, Fill, H American Elsevier Cytostatic drug therapy of thyroid cancer. in Robbins, J; Braverman, L (eds) Thyroid Research: New York.
429. Simon, D, Koehrle, J, Reiners, C, et al. 1998. Redifferentiation therapy with retinoids. therapeutic option for advanced follicular and papillary thyroid carcinoma. World J Surg 22:569-574.
430. Schmutzler, C, Kohrle, J 2000. Retinoic acid redifferentiation therapy for thyroid cancer. Thyroid 10:393-406.
431. Furuya, F; Shimura, H; Suzuki, H; Taki, K; Ohta, K; Haraguchi, K; Onaya, T; Endo, T; Kobayashi, T. Histone deacetylase inhibitors restore radioiodine uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology 2004.
432. Fortunati, N; Catalano, MG; Arena, K; Brignardello, E; Piovesan, A; Boccuzzi, G. Valproic acid induces the expression of the Na+/I- symporter and iodine uptake in poorly differentiated thyroid cancer cells. J Clin Endocrinol Metab 89:1006-1009, 2004.
433. Martelli, M L, Iuliano, R, Le Pera, I, et al. 2002 Inhibitory effects of peroxisome poliferator-activated receptor gamma on thyroid carcinoma cell growth. J Clin Endocrinol Metab 87:4728-35.
434. Portella, G, Scala, S, Vitagliano, D, Vecchio, G, Fusco, A 2002 ONYX-015, an E1B gene-defective adenovirus, induces cell death in human anaplastic thyroid carcinoma cell lines. J Clin Endocrinol Metab 87:2525-31.
435. Tanaka, K; Towata, S; Nakao, K; Mizuguchi, H; Hayakawa, T; Niwa, M; Ishii, N; Nagayama, Y. Thyroid cancer immunotherapy with retroviral and adenoviral vectors expressing granulocyte macrophage colony stimulating factor and interleukin-12 in a rat model. Clin Endocrinol 59:734-742, 2003.
436. Braga-Basaria, M; Ringel, MD. Beyond radioiodine: A review of potential new therapeutic approaches for thyroid cancer. J Clin Endocrinol Metab 88:1947-1960, 2003.
437. Brilli L, Pacini F. Targeted therapy in refractory thyroid cancer: current achievements and limitations. Future Oncol. 7:657-68, 2011.
438. Wells SA Jr, Gosnell JE, Gagel RF et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 28:767-72, 2010.
439. .Robinson BG, Paz-Ares L, Krebs A, Vasselli J, Haddad R. Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Endocrinol Metab. 95:2664-71, 2010. Giuffrida D, Prestifilippo A, Scarfia A, Martino D, Marchisotta S. New treatment in advanced thyroid cancer.J Oncol. 2012;2012:391629doi: 10.1155/2012/391629
440. Carhill AA, Cabanillas ME, Jimenez C, Waguespack SG, Habra MA, Hu M, Ying A, Vassilopoulou-Sellin R, Gagel RF, Sherman SI, Busaidy NL. The noninvestigational use of tyrosine kinase inhibitors in thyroid cancer: establishing a standard for patient safety and monitoring. J Clin Endocrinol Metab. 2013 Jan;98(1):31-42. doi: 10.1210/jc.2012-2909