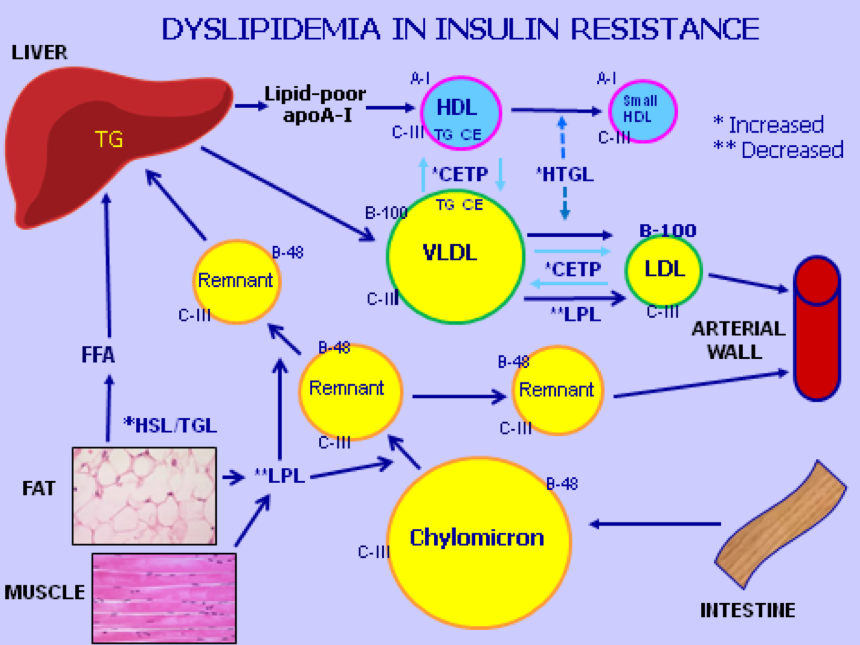
Figure 1. Lipoprotein Metabolism in Insulin Resistance: A combination of excess production and delayed disposal results in secondary HTG and atherogenic dyslipidemia in the insulin resistant state. Chylomicrons and VLDL production originating from the intestine and liver are increased. Mobilization of free fatty acids (FFA) from fat cells by hormone sensitive and TG lipases (HSL/TGL) provides the liver with substrate for VLDL formation. Dietary intake of fat provides the intestine with TG for chylomicron formation, which is upregulated in insulin resistance. Hepatic VLDL containing excess apoC-III relative to apoE is increased; apoC-III delays receptor-mediated hepatic uptake of VLDL and chylomicron remnants resulting in formation of intermediate density lipoproteins (IDL, not shown) and smaller and denser low-density lipoproteins (LDL). Lipoprotein lipase (LPL) is inhibited by apoC-III and decreased by insulin resistance and/or deficiency. Cholesterol ester transfer protein (CETP) is upregulated resulting in exchange of TG and cholesterol ester (CE), leading to TG enrichment of LDL and HDL. Both become substrates for hepatic triglyceride lipase (HTGL), which is upregulated and acts on TG-enriched HDL and LDL to make them smaller, atherogenic and dysfunctional. Apolipoproteins A-I, B-48, B-100, C-I, C-II, C-III (C), and E are labelled and play important roles in lipoprotein metabolism.