Figure 16: GR mutations GRR614Q and V423A respectively damage protein conformation of LBD and DBD, and attenuates actions of these domains.
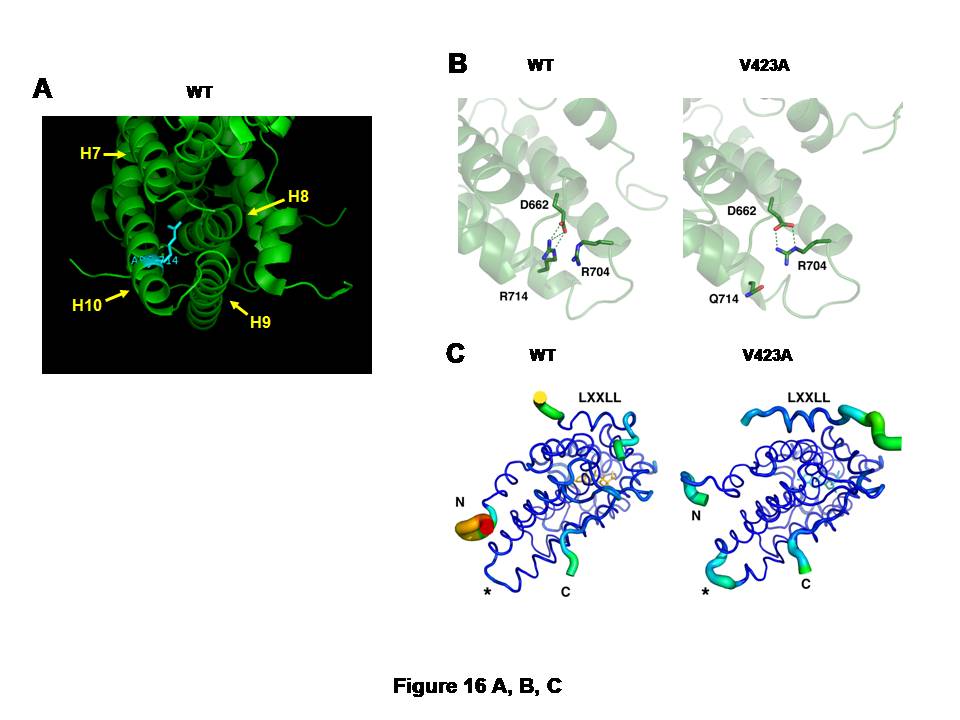
A, and CB: Arginine (R) to glutamine (Q) replacement at amino acid 714 of the hGR alters 3-dimensional structure of its LBD.
The large, positively charged side chain of arginine (R) at amino acid 714 of the hGR protrudes into the space surrounded by helices 7 to 10, suggesting its important role in holding the alignment of these -helices (A). Arginine (R) to glutamine (Q) replacement at this amino acid position causes formation of a new salt bridge between arginine (R) at 704 and aspartic acid (D) at 662 (B).In the wild type hGR LBD, R714 is tightly bound to D662 through an electrostatic salt bridge (left panel). Substitution of arginine for glutamine in the hGRR714Q LBD results in a rearrangement of the side chains, forming a new salt bridge between R704 and D662, while displacing Q714 (right panel). This relaxes some constraint on helix 10 and results in structural changes throughout the LBD.
hGRR714Q LBD has the destabilized AF-2 surface that blocks optimal binding of the LXXLL coactivator motif (C).The thickness and color of the C trace of the wild type hGR LBD (left panel) and hGRR714Q LBD (right panel) indicate the areas of least (thin and blue) to most (thick and red) motion over the course of the simulation. As expected, the termini of the LBD and the bound peptide move the most. There is significant motion at the mutation site (*) in the mutant LBD, but not in the wild type, suggesting the mutation caused destabilization in this structural area. The bound LXXLL peptide is thus more labile in the mutant LBD structure as seen by the thickness of its C trace.