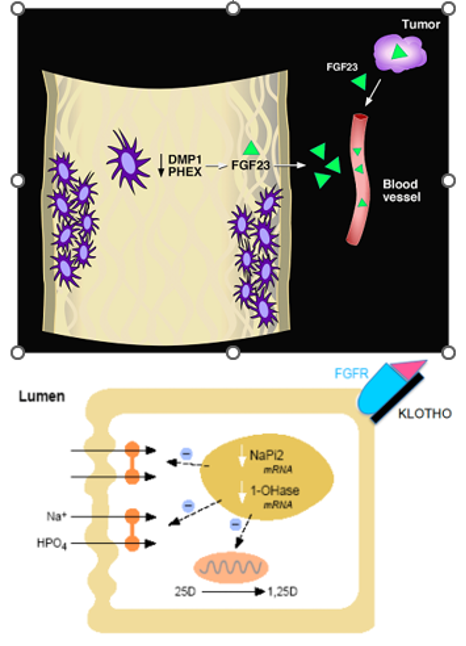
Figure 6. Scheme for the speculated pathophysiology of XLH, ARHR, TIO, and ADHR. Upper panel, osteocytes, comprising a network of connected cells embedded in mineralized bone are the cellular source of PHEX (which is mutated in XLH), DMP1 (which is mutated in ARHR), and FGF23 (which is found in high concentrations in all four of these hypophosphatemic disorders). It follows that loss of PHEX or DMP1 results in increased FGF23 production/secretion by mechanisms that are not currently understood. Circulating FGF23 concentrations may also occur secondary to the increased production associated with various tumors. Lower panel, circulating FGF23 interacts with an FGF receptor (presumably FGFR1) on the basolateral surface of the proximal renal tubular cell. Klotho, produced by the distal renal tubule in both membrane bound and secretory forms, is necessary for the FGF23/FGFR interaction. Signaling through this pathway results in a decrease in NPT2 mRNAs, thereby reducing the abundance of Pi cotransporters on the apical membrane as well as a more rapid action of translocating the transporter off of the apical membrane. Thus, impairment of renal tubular Pi reabsorption results. Likewise, synthesis of 1,25(OH)2D is impaired, while its clearance is augmented. In XLH and ARHR, increased production of FGF23 occurs in the skeleton; in TIO, increased production of FGF23 occurs in tumors; in ADHR, enhanced activity of FGF23 occurs as a result of the specific mutations that retard its metabolic clearance.